Pakar medis dalam artikel tersebut

Publikasi baru
Penyakit Glanzmann: penyebab, gejala, diagnosis, dan pengobatan
Terakhir diperbarui: 14.03.2026

Kami memiliki pedoman sumber yang ketat dan hanya menautkan ke situs medis tepercaya, lembaga penelitian akademis, dan, jika memungkinkan, studi yang telah ditinjau sejawat secara medis. Harap dicatat bahwa angka dalam tanda kurung ([1], [2], dll.) adalah tautan yang dapat diklik ke studi-studi ini.
Jika Anda merasa ada konten kami yang tidak akurat, kedaluwarsa, atau dipertanyakan, silakan pilih dan tekan Ctrl + Enter.
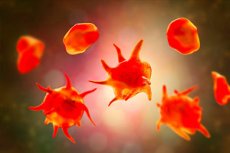
Penyakit Glanzmann, yang lebih akurat dikenal dalam hematologi sebagai trombastenia Glanzmann, adalah kelainan trombosit herediter yang langka. Pada kondisi ini, jumlah trombosit biasanya normal, tetapi trombosit itu sendiri gagal menempel dengan benar satu sama lain dan membentuk sumbatan primer lengkap di tempat cedera pembuluh darah. Hal ini menyebabkan perdarahan berulang, terutama melibatkan selaput lendir dan kulit. Tinjauan modern menggambarkan penyakit Glanzmann sebagai salah satu kelainan trombosit herediter yang paling banyak dipelajari dan kelainan trombosit herediter paling umum dengan perdarahan mukokutan yang parah. [1]
Penyakit ini didasarkan pada defisiensi kuantitatif atau defek kualitatif integrin alfa IIb beta 3 pada permukaan trombosit. Reseptor ini diperlukan oleh trombosit untuk mengikat fibrinogen dan membentuk agregat yang stabil. Ketika mekanisme ini terganggu, kontrol perdarahan menjadi lambat dan tidak stabil, bahkan dengan cedera rumah tangga biasa, prosedur gigi, atau menstruasi. Penyakit ini dimulai sejak lahir, tetapi tingkat keparahan gejalanya dapat sangat bervariasi bahkan di antara anggota keluarga dengan varian genetik yang sama. [2]
Penyakit Glanzmann secara klinis signifikan karena seringkali menyerupai penyebab perdarahan yang lebih umum, seperti penyakit von Willebrand, "pembuluh darah lemah," atau kecenderungan mimisan yang tidak spesifik. Kesalahan diagnosis dapat menyebabkan penanganan yang tidak tepat sebelum operasi, persalinan, atau perdarahan hebat. Namun, teknik diagnostik laboratorium modern, sitometri aliran, konfirmasi molekuler, dan penggunaan faktor VII aktif rekombinan yang lebih luas telah secara signifikan meningkatkan penanganan pasien ini. [3]
Dari perspektif praktis, penyakit ini tidak hanya memerlukan diagnosis tetapi juga strategi manajemen jangka panjang: menghindari obat-obatan yang tidak perlu yang mengganggu fungsi trombosit, merencanakan intervensi invasif sebelumnya, mengendalikan defisiensi zat besi, mengelola perdarahan menstruasi dengan tepat, dan terutama mempersiapkan kehamilan dan persalinan dengan hati-hati. Pada tahun 2025, survei spesialis Eropa menunjukkan bahwa bahkan dalam lingkungan khusus, pendekatan seragam untuk manajemen penyakit Glanzmann masih bervariasi, menyoroti perlunya pengobatan di pusat-pusat yang berpengalaman dalam menangani kondisi perdarahan langka. [4]
Kodekan berdasarkan ICD-10 dan ICD-11
Penyakit Glanzmann adalah kelainan kualitatif trombosit herediter. Dalam Klasifikasi Penyakit Internasional, revisi ke-10, biasanya dikodekan di bawah judul D69.1 - kelainan kualitatif trombosit. Ini adalah kode yang praktis, tetapi tidak sepenuhnya detail, karena dalam Klasifikasi Penyakit Internasional, revisi ke-10, penyakit trombosit herediter langka sering digabungkan ke dalam kategori yang lebih luas. [5]
Klasifikasi Penyakit Internasional, revisi ke-11, telah menyempurnakan pengkodean. Penyakit Glanzmann biasanya diberi kode 3B62.0Y—cacat kualitatif herediter trombosit lainnya yang ditentukan. Hal ini mencerminkan fakta klinis yang penting: penyakit ini memiliki sifat molekuler yang jelas, tetapi dalam Klasifikasi Penyakit Internasional, revisi ke-11, penyakit ini lebih sering dicantumkan dalam blok cacat kualitatif herediter trombosit, daripada sebagai kategori "umum" terpisah dengan nama independen pada tingkat pertama klasifikasi. [6]
Kode-kode utama dirangkum dalam tabel. [7]
| Nosologi | Klasifikasi Penyakit Internasional, revisi ke-10 | Klasifikasi Penyakit Internasional, revisi ke-11 |
|---|---|---|
| Penyakit Glanzmann | D69.1 | 3B62.0Y |
| Grup Status | Cacat trombosit kualitatif | Cacat kualitatif trombosit herediter |
Epidemiologi
Penyakit Glanzmann dianggap sangat langka. Perkiraan prevalensi yang paling sering dikutip adalah sekitar 1 kasus per 1.000.000 orang dalam populasi umum. Ini berarti bahwa sebagian besar dokter umum tidak akan pernah menemui pasien seperti itu, dan bahkan banyak ahli hematologi hanya melihat beberapa kasus selama karier mereka. Kelangkaan penyakit ini telah lama menghambat pengembangan pedoman klinis utama dan registri internasional yang terpadu. [8]
Distribusi penyakit di seluruh dunia tidak merata. Insidensinya secara signifikan lebih tinggi pada populasi dengan perkawinan sedarah dan efek pendiri. Contoh klasik termasuk beberapa kelompok Romani, komunitas Manoush Prancis, populasi tertentu di India Selatan, Yahudi Irak, beberapa kelompok nomaden di Yordania, serta beberapa wilayah Pakistan dan Newfoundland dan Labrador. Pada populasi tersebut, penyakit ini terdeteksi jauh lebih sering daripada yang diperkirakan berdasarkan perkiraan global secara keseluruhan. [9]
Penyakit ini menyerang pria dan wanita, karena diwariskan secara autosomal resesif. Namun, beban klinis pada wanita seringkali lebih tinggi karena pendarahan menstruasi, kehilangan darah saat melahirkan, dan komplikasi reproduksi. Menurut data registri dan tinjauan, gejala pertama sering muncul pada tahun pertama kehidupan, dan sebagian besar pasien didiagnosis pada masa kanak-kanak; menurut satu registri, 85% pasien didiagnosis pada usia 14 tahun. [10]
Pedoman epidemiologi disajikan dalam tabel. [11]
| Indikator | Landmark |
|---|---|
| Perkiraan prevalensi pada populasi umum | sekitar 1 banding 1.000.000 |
| Populasi berisiko tinggi | komunitas dengan perkawinan sedarah dan efek pendiri |
| Jenis pewarisan | autosomal resesif |
| Usia rata-rata munculnya gejala | seringkali tahun pertama kehidupan |
| Proporsi pasien yang didiagnosis pada usia 14 tahun | sekitar 85% |
Alasan
Penyebab utama penyakit Glanzmann adalah varian patogenik pada gen integrin alfa IIb, ITGA2B, atau gen integrin beta 3, ITGB3. Kedua gen tersebut terletak pada lengan panjang kromosom 17 dan mengkode subunit reseptor yang membentuk integrin alfa IIb beta 3 pada permukaan trombosit. Ketika struktur atau transportasi reseptor ini terganggu, trombosit tetap "hidup" dan ada dalam darah, tetapi kehilangan kemampuannya untuk beragregasi secara efektif. [12]
Penyakit ini berkembang ketika kedua alel dari salah satu gen ini membawa varian patogenik. Ini bisa berupa varian yang sama dari kedua orang tua, atau dua varian yang berbeda, yaitu heterozigositas majemuk. Ratusan mutasi telah dijelaskan: varian missense, varian nonsense, kelainan situs penyambungan, serta delesi dan duplikasi besar yang jarang terjadi. Dalam praktiknya, ini berarti bahwa gambaran molekuler penyakit ini sangat bervariasi, meskipun fenotipe klinisnya seringkali serupa. [13]
Penyakit Glanzmann klasik bersifat herediter, namun varian langka yang didapat juga telah dijelaskan dalam literatur. Varian ini dikaitkan dengan autoantibodi terhadap reseptor integrin alfa IIb beta 3 dan dapat terjadi pada penyakit autoimun, sel plasma, dan limfoproliferatif, serta dengan paparan obat yang memblokir reseptor ini. Varian ini penting terutama untuk diagnosis banding, karena pada orang dewasa dengan onset terlambat dan tanpa riwayat keluarga, bentuk herediter harus dipertimbangkan di luar bentuk herediter. [14]
Faktor risiko
Mengenai risiko penyakit itu sendiri, faktor utamanya adalah pewarisan dua alel patogen. Oleh karena itu, risikonya lebih tinggi pada keluarga dengan kerabat yang sudah terkena penyakit, serta pada populasi dengan frekuensi perkawinan sedarah yang tinggi. Dengan pewarisan autosomal resesif, orang tua dari individu yang terkena penyakit biasanya sehat secara klinis tetapi merupakan pembawa. Inilah sebabnya mengapa penyakit ini mungkin pertama kali muncul dalam keluarga secara tidak terduga—setelah kelahiran anak dengan pendarahan hebat. [15]
Dalam hal risiko pendarahan hebat pada pasien yang sudah sakit, periode paling berbahaya adalah masa kanak-kanak awal, menarche, kehamilan, persalinan, intervensi gigi dan bedah, trauma, dan situasi di mana pasien menerima obat-obatan yang semakin mengganggu fungsi trombosit. Aspirin, banyak obat antiinflamasi nonsteroid, dan beberapa obat lain yang memengaruhi hemostasis menimbulkan masalah khusus. [16]
Riwayat transfusi trombosit merupakan faktor risiko tambahan untuk hasil yang tidak menguntungkan. Meskipun seringkali menyelamatkan nyawa, transfusi ini meningkatkan kemungkinan berkembangnya antibodi terhadap antigen leukosit manusia dan terhadap integrin alfa IIb beta 3 itu sendiri. Hal ini dapat mengurangi efektivitas transfusi di masa mendatang, dan selama kehamilan, antibodi tersebut dapat menimbulkan risiko bagi janin dan bayi baru lahir. [17]
Faktor risiko yang penting secara praktis dirangkum dalam tabel. [18]
| Kategori risiko | Apa yang meningkatkan risikonya? |
|---|---|
| Awal mula penyakit | Riwayat keluarga, kedua orang tua adalah pembawa gen, pernikahan sedarah. |
| Pendarahan hebat | masa kanak-kanak, menarche, persalinan, operasi, cedera |
| Peningkatan perdarahan | asam asetilsalisilat, banyak obat antiinflamasi nonsteroid, dan obat-obatan lain dengan efek antiplatelet |
| Komplikasi pengobatan | transfusi trombosit berulang, aloimunisasi |
Patogenesis
Patogenesis penyakit Glanzmann dikaitkan dengan tidak adanya atau kerusakan reseptor agregasi trombosit utama, integrin alfa IIb beta 3. Biasanya, setelah aktivasi trombosit, reseptor ini mengubah konfigurasi dan mulai mengikat fibrinogen, kemudian membentuk "jembatan" antara trombosit yang berdekatan. Inilah cara sumbat trombosit primer terbentuk. Pada penyakit Glanzmann, mekanisme ini terblokir. [19]
Gangguan ini dapat bersifat kuantitatif atau kualitatif. Dalam beberapa kasus, reseptor pada permukaan sangat rendah, sedangkan pada kasus lain, reseptor tersebut ada tetapi tidak mampu mengikat fibrinogen dengan benar atau mengirimkan sinyal ke dalam sel. Karena itu, trombosit dapat mengenali kerusakan pembuluh darah dan menempel padanya sejak dini, tetapi gagal membentuk agregat yang lengkap dan stabil. Hal ini menjelaskan mengapa jumlah trombosit biasanya tidak berkurang, namun pendarahan tetap parah. [20]
Masalahnya tidak hanya terbatas pada agregasi saja. Pada penyakit Glanzmann, stabilisasi sumbat trombosit, retraksi bekuan, dan sampai batas tertentu, pembentukan trombin juga terganggu. Oleh karena itu, bahkan jika perlekatan trombosit awal telah terjadi, penghalang hemostatik yang dihasilkan tetap lebih lemah dan lebih mudah terganggu. Hal ini menjelaskan perdarahan yang berkepanjangan setelah cedera atau operasi. [21]
Menariknya, tingkat keparahan perdarahan tidak selalu sesuai dengan jumlah reseptor pada permukaan trombosit. Tinjauan modern menyoroti variabilitas klinis yang luas. Beberapa pasien dengan ekspresi reseptor yang sangat rendah memiliki gejala yang relatif ringan, sementara yang lain, sebaliknya, mengalami perdarahan hebat. Faktor vaskular dan hemostatik tambahan, serta varian genetik yang memodifikasi, kemungkinan mempengaruhi fenotipe. [22]
Gejala
Gambaran klinis klasik penyakit Glanzmann adalah perdarahan mukokutan. Gejala yang paling umum meliputi mimisan, gusi berdarah, mudah memar, petekia, dan purpura. Pada anak-anak, mimisan seringkali merupakan gejala pertama dan paling terlihat, sehingga mendorong dilakukannya pemeriksaan. [23]
Pada anak perempuan dan wanita, pendarahan menstruasi yang berat sangatlah signifikan. Menorrhagia adalah salah satu manifestasi penyakit Glanzmann yang paling umum dan parah secara klinis. Hal ini bisa sangat parah sehingga menyebabkan rawat inap, transfusi, dan anemia defisiensi besi kronis, dan pada awal menarche, hal ini bisa menjadi manifestasi dramatis pertama dari penyakit tersebut. [24]
Perdarahan setelah cedera dan prosedur juga umum terjadi. Pasien sering mengalami perdarahan berkepanjangan setelah pencabutan gigi, operasi, sunat, trauma pada mukosa mulut, dan bahkan setelah prosedur yang relatif ringan. Perdarahan saluran pencernaan kurang umum tetapi signifikan secara klinis; menurut sumber genetik resmi Perpustakaan Kedokteran Nasional AS, sekitar seperempat pasien mengalaminya pada suatu saat dalam hidup mereka. [25]
Perdarahan intrakranial dan hemartrosis telah dijelaskan, namun jarang terjadi. Menurut tinjauan terbaru, perdarahan gastrointestinal terjadi pada sekitar 10%-20% pasien, dan perdarahan intrakranial pada 1%-2%. Komplikasi ini jarang terjadi namun paling berbahaya, karena dikaitkan dengan risiko kecacatan dan kematian tertinggi. [26]
Seiring bertambahnya usia, perdarahan spontan mungkin menjadi kurang sering terjadi pada beberapa pasien, meskipun tidak sepenuhnya hilang. Hal ini terutama terlihat pada mimisan. Namun, perdarahan menstruasi dan obstetri, sebaliknya, tetap menjadi masalah utama pada wanita usia reproduksi, dan intervensi bedah berbahaya pada usia berapa pun. [27]
Manifestasi klinis utama dirangkum dalam tabel. [28]
| Manifestasi | Perkiraan frekuensi atau nilai |
|---|---|
| Mimisan | sangat umum, terutama pada masa kanak-kanak |
| Gusi berdarah | manifestasi yang sering terjadi |
| Menorrhagia | manifestasi yang sangat umum dan signifikan secara klinis |
| Petechiae, purpura, memar | khas |
| Pendarahan saluran pencernaan | sekitar 10%-20%, menurut beberapa sumber hingga seperempat pasien selama masa hidup mereka. |
| Perdarahan intrakranial | langka, tetapi paling berbahaya |
| Perdarahan pasca operasi dan pasca trauma | ciri |
Klasifikasi, bentuk, dan tahapan
Klasifikasi klasik penyakit Glanzmann didasarkan pada jumlah dan fungsi integrin alfa IIb beta 3 pada permukaan trombosit. Tipe I ditandai dengan ekspresi reseptor kurang dari 5% dari normal. Tipe II biasanya diekspresikan pada tingkat 5%-20% dari normal. Pada bentuk varian, reseptor hadir dalam jumlah yang cukup tetapi secara fungsional cacat dan tidak mendukung agregasi normal. [29]
Dari sudut pandang praktis, klasifikasi ini berguna tetapi tidak sempurna. Klasifikasi ini menggambarkan fenotipe laboratorium dengan baik, tetapi tidak selalu secara akurat memprediksi tingkat keparahan perdarahan. Oleh karena itu, di klinik, fenotipe perdarahan sebenarnya dinilai lebih lanjut: seberapa sering mimisan terjadi, apakah ada perdarahan menstruasi yang berat, apakah diperlukan transfusi, dan apakah ada episode pasca operasi atau obstetri yang berbahaya. [30]
Penyakit Glanzmann tidak memiliki sistem penentuan stadium universal yang ketat, seperti halnya tumor atau gagal jantung kronis. Dokter biasanya merujuk pada perjalanan klinis ringan, sedang, dan berat berdasarkan frekuensi dan lokasi perdarahan, kebutuhan rawat inap, perkembangan anemia, alloimunisasi, dan kebutuhan akan agen hemostatik khusus. Oleh karena itu, bagian "stadium" untuk penyakit ini lebih tepat dipahami sebagai tingkat keparahan klinis, daripada penentuan stadium internasional yang kaku. [31]
Klasifikasi disajikan dengan mudah dalam tabel. [32]
| Pilihan | Ciri |
|---|---|
| Tipe I | kurang dari 5% dari ekspresi normal integrin alfa IIb beta 3 |
| Tipe II | sekitar 5%-20% dari ekspresi normal |
| Bentuk varian | Jumlah reseptornya mencukupi, tetapi fungsinya cacat. |
| Tingkat keparahan klinis | dinilai berdasarkan frekuensi dan tingkat keparahan pendarahan, bukan berdasarkan panggung internasional. |
Komplikasi dan konsekuensi
Komplikasi jangka panjang yang paling umum adalah defisiensi zat besi kronis dan anemia defisiensi zat besi. Hal ini berkembang dengan latar belakang mimisan berulang, pendarahan gusi, pendarahan saluran pencernaan, dan terutama pendarahan menstruasi. Pada anak-anak, hal ini dapat bermanifestasi sebagai kelemahan dan penurunan aktivitas fisik, sedangkan pada orang dewasa, dapat mengakibatkan kelelahan, penurunan produktivitas, dan penurunan kualitas hidup. [33]
Komplikasi yang paling berbahaya adalah perdarahan hebat setelah operasi, trauma, persalinan, dan perdarahan intrakranial yang jarang terjadi. Bagi wanita, kehilangan darah obstetri yang parah dan masalah yang terkait dengan antibodi trombosit merupakan kelompok risiko tersendiri. Tinjauan tahun 2024 tentang kehamilan pada penyakit Glanzmann menekankan risiko tinggi perdarahan ibu dan janin, dengan insiden perdarahan pasca persalinan dalam seri yang dipublikasikan mencapai 40%-50%. [34]
Sebagian besar komplikasi tidak begitu berkaitan dengan penyakit itu sendiri, melainkan dengan pengobatannya. Transfusi trombosit berulang dapat menyebabkan pembentukan antibodi terhadap antigen leukosit manusia dan integrin alfa IIb beta 3, sehingga transfusi selanjutnya menjadi kurang efektif. Menurut tinjauan, hingga 30% pasien dapat mengembangkan antibodi tersebut, dan ketika menggunakan trombosit yang dikurangi leukositnya, antigen leukosit manusia masih berkembang pada sekitar 17% pasien. [35]
Kapan harus menemui dokter?
Konsultasikan dengan dokter jika Anda mengalami mimisan berulang, perdarahan berkepanjangan setelah cedera ringan, perdarahan setelah pencabutan gigi, memar yang sering tanpa sebab yang jelas, perdarahan menstruasi yang signifikan, atau riwayat keluarga dengan gangguan perdarahan langka. Pada anak-anak, episode perdarahan yang sering terjadi pada usia dini dan perdarahan yang sangat banyak setelah cedera umum pada masa kanak-kanak atau sunat sangat mencurigakan. [36]
Perhatian medis segera diperlukan untuk pendarahan hebat yang terus menerus, muntah darah, feses hitam, kelemahan parah, tanda-tanda anemia setelah kehilangan darah, dugaan pendarahan intrakranial, pendarahan selama kehamilan, dan sebelum operasi darurat. Untuk pasien yang sudah didiagnosis, prosedur invasif apa pun harus direncanakan terlebih dahulu daripada dilakukan secara mendadak, karena tindakan lokal rutin mungkin tidak cukup. [37]
Alasan lain untuk konsultasi dini adalah persiapan kehamilan dan persalinan. Tinjauan modern merekomendasikan agar wanita tersebut menerima pemantauan di pusat khusus, penilaian antibodi, konseling reproduksi, dan perencanaan manajemen hemostatik sebelum pembuahan. Hal ini membantu mengurangi risiko perdarahan postpartum yang parah dan komplikasi janin. [38]
Alasan-alasan terpenting untuk menghubungi dokter tercantum dalam tabel. [39]
| Situasi | Mengapa ini penting? |
|---|---|
| Mimisan berulang sejak kecil | Kemungkinan adanya kelainan bawaan pada fungsi trombosit adalah suatu kemungkinan. |
| Pendarahan setelah pencabutan gigi atau operasi kecil | pemicu klinis tipikal untuk deteksi penyakit |
| Menstruasi yang sangat deras | risiko tinggi anemia dan rawat inap |
| Operasi elektif, persalinan, prosedur invasif | Persiapan hemostatik awal diperlukan. |
| Pendarahan gastrointestinal, intrakranial, atau masif. | kondisi darurat |
Diagnostik
Diagnosis penyakit Glanzmann selalu dimulai dengan kecurigaan klinis. Dokter mengevaluasi sifat perdarahan, usia onset, riwayat keluarga, adanya kehilangan darah yang parah setelah prosedur gigi dan bedah, dan, pada wanita, riwayat menstruasi dan obstetri. Bahkan pada tahap ini, penting untuk memahami ciri utama: pada penyakit Glanzmann, jumlah trombosit biasanya normal, dan masalahnya terletak pada fungsinya. [40]
Langkah laboratorium pertama meliputi hitung darah lengkap, apusan darah tepi, dan tes koagulasi dasar. Jumlah dan ukuran trombosit biasanya normal, begitu pula waktu protrombin dan waktu tromboplastin parsial teraktivasi. Jika anemia terjadi, biasanya disebabkan oleh kehilangan darah kronis. Jika trombositopenia, makrotrombositosis yang nyata, atau kelainan koagulasi yang parah terdeteksi secara bersamaan, diagnosis lain harus dipertimbangkan. [41]
Langkah selanjutnya adalah menyingkirkan penyebab perdarahan yang lebih umum, terutama penyakit von Willebrand. Oleh karena itu, sesuai rekomendasi, antigen faktor von Willebrand, aktivitas terkait ristocetin, dan aktivitas faktor VIII dinilai. Tes-tes ini biasanya normal pada penyakit Glanzmann, dan ini membantu mempersempit pencarian. [42]
Standar emas untuk diagnosis adalah agregometri transmisi cahaya. Penyakit Glanzmann ditandai dengan profil yang hampir patognomonik: agregasi yang sangat berkurang atau tidak ada terhadap semua agonis fisiologis dengan aglutinasi yang tetap terjaga terhadap ristocetin. Hasil ini sebaiknya dikonfirmasi dengan tes ulang, karena tes ini sangat dipengaruhi oleh kondisi pra-analitik. [43]
Sitometri aliran yang menilai ekspresi integrin alfa IIb beta 3 pada permukaan trombosit sangat penting. Hal ini memungkinkan konfirmasi cepat defisiensi reseptor kuantitatif dan penentuan jenis penyakit, tetapi memiliki keterbatasan: pada bentuk varian, reseptor mungkin diekspresikan hampir normal, namun masalahnya tetap fungsional. Oleh karena itu, ekspresi normal tidak mengecualikan bentuk varian dan harus dibandingkan dengan agregometri. [44]
Tes tambahan meliputi penilaian retraksi bekuan darah, yang biasanya berkurang pada penyakit Glanzmann, serta konfirmasi genetik molekuler. Pengujian genetik berguna tidak hanya untuk verifikasi diagnosis akhir tetapi juga untuk identifikasi pembawa, konseling keluarga, dan diagnosis prenatal dan praimplantasi. Dengan fenotipe laboratorium yang khas, logis untuk menganalisis gen ITGA2B dan ITGB3 terlebih dahulu. Jika sekuensing standar gagal mendeteksi kedua mutasi patogenik, pertimbangkan untuk mencari delesi dan duplikasi. [45]
Diagnosis instrumental untuk penyakit Glanzmann bukanlah cara utama untuk mengkonfirmasi defek trombosit, tetapi banyak digunakan bila diperlukan. Pemeriksaan oleh dokter spesialis THT diperlukan untuk mimisan yang terus-menerus, endoskopi untuk perdarahan gastrointestinal, USG panggul untuk menoragia berat, dan, jika terdapat gejala neurologis, pencitraan neurologis mendesak untuk menyingkirkan kemungkinan perdarahan intrakranial. Dengan demikian, bagian "instrumental" dari diagnosis di sini tidak ditujukan pada mekanisme penyakit itu sendiri, tetapi pada lokasi dan konsekuensi dari perdarahan. [46]
Algoritma diagnostik langkah demi langkah disajikan dalam tabel. [47]
| Panggung | Apa yang sedang dinilai? | Apa yang dapat diharapkan dari penyakit Glanzmann? |
|---|---|---|
| 1 | Riwayat pendarahan dan riwayat keluarga | Perdarahan mukokutan yang terjadi pada usia dini |
| 2 | Tes darah umum dan apusan | biasanya jumlah dan ukuran trombosit normal |
| 3 | Tes koagulasi | biasanya normal |
| 4 | Pengecualian penyakit von Willebrand | Tes biasanya tidak menunjukkan penyimpangan karakteristik. |
| 5 | Agregometri transmisi cahaya | Tidak ada agregasi terhadap sebagian besar agonis, respons normal terhadap ristocetin. |
| 6 | Sitometri aliran | penurunan atau perubahan ekspresi integrin alfa IIb beta 3 |
| 7 | Tes genetik | varian patogenik pada ITGA2B atau ITGB3 |
Diagnosis banding
Dalam praktiknya, penyakit Glanzmann paling sering harus dibedakan dari penyakit von Willebrand dan gangguan fungsi trombosit bawaan lainnya. Penyakit von Willebrand jauh lebih umum dan juga menyebabkan mimisan, menoragia, dan perdarahan pasca prosedur. Oleh karena itu, pengukuran faktor von Willebrand normal dan profil agregometri yang khas sangat penting. [48]
Diagnosis bersamaan yang sangat penting adalah sindrom Bernard-Soulier. Secara klinis, gejalanya bisa serupa, tetapi biasanya ditandai dengan makrotrombositopenia, dan agregometri secara spesifik mendeteksi respons ristocetin yang abnormal, sedangkan pada penyakit Glanzmann, aglutinasi ristocetin tetap terjaga. Flow cytometry memberikan dukungan tambahan, karena kompleks reseptor yang berbeda terpengaruh pada sindrom Bernard-Soulier. [49]
Kelompok penting berikutnya adalah kelainan agregasi bawaan langka lainnya, termasuk defisiensi kindlin 3 pada defisiensi adhesi leukosit tipe III dan defek pada gen RASGRP2. Pada defisiensi adhesi leukosit tipe III, profil agregasi mungkin menyerupai bentuk varian penyakit Glanzmann, tetapi pasien juga akan mengalami infeksi bakteri yang sering dan gangguan penyembuhan luka. Pada defek RASGRP2, profil agregometri biasanya tidak sepenuhnya konsisten dan memerlukan analisis genetik yang lebih luas. [50]
Penyakit Glanzmann yang didapat tidak boleh diabaikan. Onset yang terlambat pada orang dewasa, tidak adanya riwayat masa kanak-kanak yang khas, adanya gangguan autoimun, sel plasma, atau limfoproliferatif, dan hubungan dengan obat-obatan yang memblokir integrin alfa IIb beta 3 harus menimbulkan kecurigaan terhadap varian yang didapat. Pasien tersebut mungkin menunjukkan gejala yang sangat mirip, tetapi penyebab dan pengobatannya akan berbeda. [51]
Terakhir, diagnosis banding mencakup disfungsi trombosit akibat obat, terutama karena asam asetilsalisilat dan banyak obat antiinflamasi nonsteroid, serta sindrom trombosit yang lebih jarang terjadi. Oleh karena itu, diagnosis tidak pernah dibuat berdasarkan satu gejala atau tes saja, tetapi hanya setelah penilaian komprehensif terhadap fenotipe, agregometri, sitometri aliran, dan genetika. [52]
Perbedaan praktis dirangkum dalam tabel. [53]
| Negara | Apa yang membantu membedakannya dari penyakit Glanzmann? |
|---|---|
| Penyakit Von Willebrand | perubahan pada faktor von Willebrand, profil tes yang berbeda |
| Sindrom Bernard-Soulier | makrotrombositopenia, gangguan respons terhadap ristocetin |
| Defisiensi adhesi leukosit tipe III | infeksi, penyembuhan luka yang buruk, ekspresi reseptor normal dengan defek fungsional |
| Cacat RASGRP2 | Untuk spektrum gangguan agregasi yang berbeda, diperlukan analisis genetik. |
| Penyakit Glanzmann yang didapat | onset terlambat, penyakit autoimun atau tumor, riwayat pengobatan |
| Disfungsi trombosit akibat obat | interaksi dengan asam asetilsalisilat, banyak obat antiinflamasi nonsteroid dan agen antiplatelet |
Perlakuan
Pengobatan penyakit Glanzmann didasarkan pada tiga tujuan: menghentikan perdarahan yang terjadi dengan segera, melakukan prosedur atau operasi dengan aman, dan meminimalkan kejadian episode parah di masa mendatang. Karena penyakit ini jarang terjadi dan secara klinis heterogen, manajemen optimal biasanya membutuhkan tim yang terdiri dari ahli hematologi, spesialis transfusi, ahli bedah, dokter gigi, ahli THT, dokter kandungan/ginekolog, dan, jika perlu, ahli genetika. Tinjauan terbaru dan survei spesialis di Eropa menekankan bahwa pengobatan harus diindividualisasi dan dilakukan di tempat yang memiliki keahlian dalam gangguan trombosit langka. [54]
Untuk perdarahan ringan, langkah pertama adalah tekanan lokal, kompres dingin, hemostasis lokal, tamponade, dan, jika perlu, koagulasi atau penjahitan. Untuk perdarahan dari gusi dan mukosa mulut, obat antifibrinolitik sering digunakan, terutama asam traneksamat, termasuk aplikasi topikal. Hal ini sangat berguna untuk prosedur gigi dan perdarahan mukosa ringan, ketika tidak perlu segera menggunakan metode sistemik dan transfusi. [55]
Terapi antifibrinolitik tetap menjadi komponen penting pengobatan bahkan untuk perdarahan sedang. Terapi ini membantu menstabilkan pembentukan bekuan darah dan sangat efektif pada rongga mulut, nasofaring, dan perdarahan menstruasi. Namun, terdapat keterbatasan: antifibrinolitik digunakan dengan hati-hati pada perdarahan saluran kemih karena risiko pembentukan bekuan darah di saluran kemih. Oleh karena itu, bahkan metode yang tampaknya "lembut" ini harus dipilih dengan mempertimbangkan lokasi perdarahan. [56]
Transfusi trombosit secara historis tetap menjadi pengobatan standar untuk perdarahan sedang hingga berat, serta profilaksis selama operasi besar. Trombosit dari satu donor, dikurangi leukosit dan, jika memungkinkan, kompatibel dengan antigen leukosit manusia, lebih disukai. Strategi ini bukan sekadar formalitas, tetapi untuk mengurangi risiko aloimunisasi dan resistensi transfusi di masa mendatang. Kehati-hatian khusus dilakukan dengan transfusi pada wanita usia reproduksi, karena antibodi yang dihasilkan dapat mempersulit kehamilan di masa mendatang. [57]
Transfusi trombosit tidak boleh menjadi respons rutin terhadap setiap episode perdarahan. Semakin besar beban transfusi, semakin tinggi risiko terbentuknya antibodi terhadap antigen leukosit manusia dan integrin alfa IIb beta 3 itu sendiri. Setelah itu, efektivitas transfusi selanjutnya menurun, dan dalam kasus yang paling parah, pasien menjadi resisten terhadap trombosit. Oleh karena itu, publikasi saat ini merekomendasikan untuk menyimpan transfusi hanya untuk situasi yang benar-benar signifikan: operasi besar, perdarahan yang mengancam jiwa, dan episode parah yang tidak merespons terapi lokal dan antifibrinolitik. [58]
Salah satu kemajuan terpenting dalam pengobatan adalah diperkenalkannya faktor VII aktif rekombinan. Faktor VII telah disetujui oleh Badan Obat-obatan Eropa dan Badan Pengawas Obat dan Makanan AS untuk pengobatan pasien dengan penyakit Glanzmann dan telah secara signifikan mengubah lanskap terapi, khususnya pada mereka yang tidak merespons dengan baik terhadap transfusi trombosit. Regimen tipikal untuk perdarahan akut, seperti yang telah ditinjau, adalah sekitar 90 mikrogram per kilogram secara intravena setiap 2-6 jam sampai hemostasis tercapai, biasanya setidaknya 3 dosis; selama operasi, obat diberikan segera sebelum prosedur dan kemudian diulang sesuai protokol. [59]
Peran obat ini sangat penting dalam keberadaan antibodi dan resistensi transfusi trombosit, tetapi penggunaannya lebih luas dalam praktik klinis dunia nyata. Registri Penyakit Glanzmann Internasional telah menunjukkan efikasi tinggi pada perdarahan non-bedah dan bedah. Untuk episode non-bedah, hemostasis yang berhasil dengan monoterapi faktor VII aktif rekombinan diamati pada sekitar 91% kasus, dan pada intervensi bedah, obat ini juga menunjukkan kontrol perdarahan yang efektif dengan insiden kejadian buruk serius yang rendah. [60]
Area pengobatan terpisah adalah pendarahan menstruasi dan kesehatan reproduksi. Anak perempuan dan wanita seringkali membutuhkan tidak hanya terapi antifibrinolitik tetapi juga koreksi hormonal untuk mengurangi volume pendarahan menstruasi. Tinjauan menjelaskan rejimen terapi hormonal berkelanjutan, penggunaan estrogen dosis tinggi dalam situasi akut, dan pada pasien yang tidak merencanakan kehamilan, ablasi endometrium atau histerektomi dibahas dalam beberapa kasus sebagai tindakan yang diperlukan untuk kehilangan darah yang refrakter. Penanganan pasien tersebut harus selalu merupakan upaya kolaboratif—seorang ahli hematologi dan seorang ahli ginekologi. [61]
Kehamilan dan persalinan memerlukan penanganan khusus. Pasien harus dipantau di pusat khusus, menjalani penilaian antibodi, risiko perdarahan ibu dan janin, rencana manajemen nyeri, dan pemilihan komponen darah. Dukungan hemostatik yang disesuaikan secara individual digunakan selama dan setelah persalinan, termasuk faktor VII aktif rekombinan, trombosit yang dikurangi leukosit, sebaiknya kompatibel dengan antigen leukosit manusia, dan asam traneksamat; rekomendasi terbaru menyarankan untuk melanjutkan dukungan antifibrinolitik setidaknya selama 3 minggu pasca persalinan. Anestesi neuraksial umumnya dikontraindikasikan. [62]
Satu-satunya pilihan pengobatan yang berpotensi radikal saat ini adalah transplantasi sel induk hematopoietik alogenik. Transplantasi ini dapat memberikan kesembuhan jangka panjang atau permanen, namun dikaitkan dengan risiko yang signifikan dan oleh karena itu hanya dipertimbangkan pada pasien yang dipilih secara cermat dengan penyakit yang sangat parah, kambuh, melumpuhkan, dan kualitas hidup yang buruk. Publikasi terbaru mengkonfirmasi bahwa transplantasi yang berhasil dimungkinkan, termasuk pada anak-anak; namun, ini bukan standar lini pertama, melainkan cadangan untuk kasus yang sangat parah. [63]
Di antara metode-metode baru, profilaksis biologis dan terapi gen adalah yang paling menarik. Tinjauan tahun 2025 menyoroti potensi antibodi monoklonal yang mempercepat pembentukan fibrin lokal, serta peran masa depan terapi lentivirus dan pengeditan gen. Kandidat profilaksis yang paling maju adalah antibodi bispesifik HMB-001, yang dirancang untuk mengakumulasi faktor VIIa endogen dan menargetkannya ke trombosit yang diaktifkan; penelitian praklinis di Nature Cardiovascular Research telah menunjukkan peningkatan pembentukan fibrin pada model penyakit Glanzmann, dan uji klinis fase 1 dan 2 sedang berlangsung di ClinicalTrials.gov. Namun, ini belum menjadi praktik standar, tetapi merupakan arah yang menjanjikan. [64]
Pendekatan pengobatan utama diringkas dengan mudah dalam tabel. [65]
| Situasi | Pendekatan dasar |
|---|---|
| Pendarahan lokal ringan | tekanan lokal, dingin, hemostasis lokal |
| Pendarahan lendir, kedokteran gigi | asam traneksamat, tindakan lokal |
| Pendarahan hebat | trombosit dan/atau faktor VII aktif rekombinan |
| Operasi | dukungan hemostatik yang direncanakan sebelumnya |
| Resistensi trombosit, antibodi | faktor VII teraktivasi rekombinan memiliki peran yang sangat penting. |
| Menorrhagia | antifibrinolitik, terapi hormonal, metode invasif jika diperlukan |
| Kehamilan dan persalinan | manajemen di pusat khusus, skema dukungan individu |
| Kursus yang sangat berat | pertimbangan transplantasi sel induk hematopoietik alogenik |
| Arah masa depan | antibodi bispesifik profilaksis, terapi gen |
Pencegahan
Pencegahan primer penyakit Glanzmann, suatu kelainan genetik, hanya dimungkinkan melalui konseling genetik. Jika sudah ada pasien dalam keluarga, serta pada populasi yang memiliki hubungan kekerabatan, deteksi pembawa, diskusi tentang risiko reproduksi, dan, jika perlu, diagnosis prenatal atau pra-implantasi bermanfaat. Analisis genetik dalam konteks ini penting tidak hanya untuk mendiagnosis pasien individu tetapi juga untuk pekerjaan pencegahan dengan keluarga. [66]
Pencegahan sekunder komplikasi jauh lebih penting dalam kehidupan sehari-hari. Pasien harus menghindari obat-obatan yang mengganggu fungsi trombosit, menjaga kebersihan mulut yang baik, segera mengobati kekurangan zat besi, memiliki rekam medis atau kartu peringatan, dan, jika memungkinkan, dipantau di pusat yang memahami gangguan trombosit herediter. Tindakan tersebut tidak menghilangkan cacat, tetapi mengurangi frekuensi episode berbahaya. [67]
Profilaksis pra-prosedural sangat penting. Setiap pencabutan gigi, operasi, endoskopi, persalinan, atau bahkan beberapa prosedur diagnostik invasif harus didiskusikan terlebih dahulu. Bagi wanita, penting untuk merencanakan menarche, masalah menstruasi, kontrasepsi, dan kehamilan jauh-jauh hari. Pasien dengan riwayat transfusi memerlukan pengujian antibodi dan pemilihan komponen trombosit yang cermat. [68]
Ramalan
Penyakit Glanzmann adalah gangguan perdarahan herediter yang parah, dan perdarahan spontan, pasca operasi, obstetri, dan pasca trauma dapat mengancam jiwa. Namun, prognosisnya tidak selalu buruk. Tinjauan terbaru mencatat bahwa dengan perawatan yang terkoordinasi dengan baik, ketersediaan faktor VII aktif rekombinan, manajemen transfusi yang tepat, dan persiapan bedah, banyak pasien mengalami kelangsungan hidup yang panjang dan relatif stabil. [69]
Tingkat keparahan penyakit ini sangat bervariasi. Pada beberapa pasien, frekuensi mimisan parah menurun seiring bertambahnya usia, namun menoragia, perdarahan pasca persalinan, dan episode pasca operasi tetap menjadi masalah klinis utama. Tidak hanya cacat genetik itu sendiri, tetapi juga anemia, alloimunisasi, akses ke pusat spesialis, dan kecepatan memulai pengobatan yang tepat memainkan peran penting dalam prognosis. [70]
Untuk sebagian kecil pasien dengan perjalanan penyakit yang sangat parah dan melumpuhkan, pendekatan radikal—transplantasi sel induk hematopoietik—dimungkinkan. Untuk sisanya, dasar prognosis yang baik adalah diagnosis dini, edukasi pasien, rencana manajemen perdarahan individual, dan persiapan yang cermat untuk semua prosedur. Dengan kata lain, penyakit ini tetap seumur hidup, tetapi dengan perawatan modern, prognosisnya jauh lebih baik daripada di era ketika pilihan terbatas pada transfusi trombosit. [71]
Pertanyaan yang Sering Diajukan (FAQ)
Apakah penyakit Glanzmann merupakan penyakit dengan jumlah trombosit rendah?
Tidak. Seringkali, jumlah trombosit normal. Masalahnya adalah trombosit gagal menggumpal secara normal karena cacat pada integrin alfa IIb beta 3. [72]
Apakah ini penyakit keturunan atau dapat muncul pada usia dewasa?
Penyakit Glanzmann klasik bersifat keturunan dan biasanya bermanifestasi pada masa bayi. Namun, terdapat varian langka yang didapat yang terkait dengan autoantibodi atau obat-obatan tertentu, sehingga timbulnya penyakit pada usia dewasa memerlukan evaluasi terpisah. [73]
Tes apa yang paling penting untuk mengkonfirmasi diagnosis?
Yang paling penting adalah agregometri transmisi cahaya, sitometri aliran, dan konfirmasi genetik molekuler. Profil laboratorium klasik adalah tidak adanya agregasi terhadap sebagian besar agonis fisiologis dengan respons yang terjaga terhadap ristocetin. [74]
Apakah penyakit ini dapat disembuhkan sepenuhnya?
Bagi sebagian besar pasien, pengobatan ditujukan untuk mencegah dan mengendalikan pendarahan, bukan untuk menghilangkan penyakit sepenuhnya. Transplantasi sel induk hematopoietik alogenik tetap menjadi pengobatan yang berpotensi radikal, tetapi hanya cocok untuk sejumlah pasien terbatas dengan kasus yang sangat parah. [75]
Mengapa transfusi trombosit tidak selalu efektif?
Karena transfusi berulang dapat menyebabkan pembentukan antibodi terhadap antigen leukosit manusia dan integrin alfa IIb beta 3. Hal ini membuat terapi trombosit kurang efektif dan menyebabkan resistensi. [76]
Apa yang dianggap sebagai kemajuan medis utama saat ini?
Salah satu kemajuan utama adalah penggunaan faktor VII aktif rekombinan, yang sangat penting dalam pendarahan hebat, pembedahan, dan respons trombosit yang buruk. Antibodi bispesifik profilaksis, seperti HMB-001, sedang dipertimbangkan sebagai arah masa depan. [77]
Apakah wanita dengan penyakit Glanzmann dapat hamil?
Ya, tetapi kehamilan dan persalinan dianggap sebagai situasi perdarahan berisiko tinggi dan harus ditangani di pusat khusus dengan pendekatan yang terencana dengan baik. Penilaian antibodi, perencanaan persalinan, dan dukungan hemostatik pada periode pascapersalinan sangat penting. [78]
Poin-poin penting dari para ahli
Alain T. Nurden, Direktur Emeritus Penelitian di Pusat Penelitian Ilmiah Nasional Prancis, Institut Hospitalo-Universitaire LIRYC, adalah salah satu peneliti penyakit Glanzmann yang paling terkenal.
Tinjauannya saat ini menekankan tiga poin utama: penyakit Glanzmann tetap menjadi model gangguan trombosit, tingkat keparahan klinis tidak selalu berkorelasi langsung dengan jenis defek laboratorium, dan masa depan pengobatan tidak hanya terletak pada pengobatan episode perdarahan individual tetapi juga pada teknologi biologis preventif, transplantasi, dan terapi gen. Dalam praktiknya, ini berarti bahwa pasien tidak dapat dinilai hanya berdasarkan jumlah sel reseptor atau mutasi tunggal. [79]
Barry S. Koller, MD, adalah Profesor Kedokteran David Rockefeller dan Direktur Laboratorium Biologi Darah dan Vaskular di Universitas Rockefeller.
Karya dan tinjauannya tentang kehamilan pada penyakit Glanzmann menyoroti aspek penting lainnya: untuk penyakit ini, sangat penting tidak hanya untuk menghentikan perdarahan tetapi juga untuk mengelola risiko alloimunisasi, resistensi transfusi, dan komplikasi janin dengan benar. Hal ini mengarah pada kesimpulan praktis: kehamilan, persalinan, dan intervensi besar apa pun harus dikelola bukan menurut pola perdarahan umum, tetapi menurut protokol yang dirancang secara khusus. [80]
Rémi Favier, MD, seorang ahli hematologi anak, adalah salah satu pendiri Pusat Rujukan Prancis untuk Gangguan Trombosit Herediter.
Posisi klinisnya, yang tercermin dalam materi pendidikan Eropa tentang gangguan trombosit herediter, selaras dengan pendekatan internasional saat ini: pasien dengan kelainan trombosit langka harus ditangani dalam jaringan khusus dengan akses ke verifikasi laboratorium, dukungan transfusi, dan pengalaman dalam skenario obstetri dan bedah yang langka. Hal ini sangat penting untuk penyakit Glanzmann, karena keputusan pengobatan sangat bergantung pada konteks dan kualitas koordinasi antar spesialis. [81]