Ahli medis artikel

Publikasi baru
Sindrom Aper
Last reviewed: 04.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.

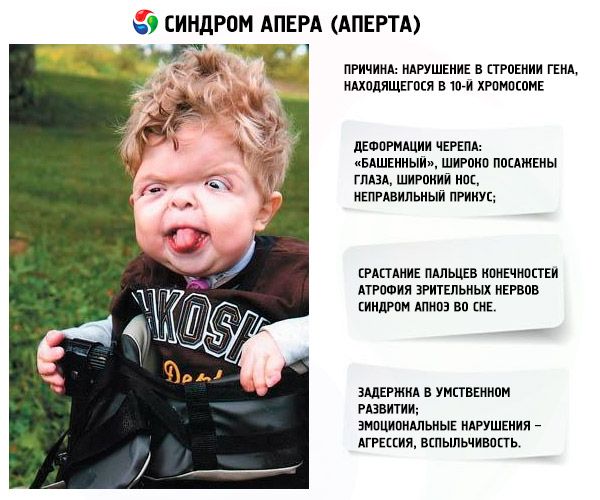
Sindrom Apert merupakan kelainan genetika yang menyebabkan cacat perkembangan pada tengkorak (mata terlalu lebar, tengkorak terlalu tinggi), dan sebagai tambahan, pada anggota tubuh bagian atas dan bawah (jari-jari pada anggota tubuh tersebut menyatu sepenuhnya; jari-jari tambahan mungkin juga muncul).
 [
[ Penyebab Sindrom Aper
Perkembangan sindrom ini dipicu oleh kelainan pada struktur gen yang terletak pada kromosom 10. Gen ini bertanggung jawab atas proses pemisahan jari, dan juga untuk penutupan sutura kranial yang tepat waktu.
Selain itu, penyakit menular yang diderita selama kehamilan (seperti rubella atau tuberkulosis, sifilis atau flu, serta meningitis), dan paparan sinar-X pada ibu dianggap sebagai penyebab patologi. Paling sering, sindrom semacam itu terdeteksi pada anak-anak yang lahir dari orang tua yang sudah lanjut usia.
Patogenesis
Sindrom Apert bersifat turun-temurun. Jenisnya adalah dominan autosomal (yaitu, dalam keluarga di mana salah satu calon orang tua sakit, kemungkinan memiliki bayi dengan penyakit ini adalah 50-100%).
Mutasi unik pada reseptor faktor pertumbuhan fibroblast 2 (FGFR2) mengakibatkan peningkatan jumlah sel progenitor yang berkembang di sepanjang jalur osteogenik. Hal ini pada akhirnya mengakibatkan peningkatan pembentukan matriks tulang subperiosteal dan osifikasi dini pada sutura kranial selama perkembangan janin. Urutan dan laju pencairan sutura menentukan tingkat deformitas dan disabilitas. Setelah bahan sutura sembuh, pertumbuhan jaringan lain yang tegak lurus terhadap sutura ini terbatas, dan tulang yang menyatu bertindak sebagai perancah tulang tunggal.
Bukti genetik pertama bahwa sindaktili pada sindrom Apert disebabkan oleh cacat pada reseptor faktor pertumbuhan keratinosit (KGFR) adalah pengamatan korelasi antara ekspresi KGFR dalam fibroblas dan tingkat keparahan sindaktili.
Ambliopia dan strabismus lebih umum terjadi pada pasien dengan mutasi FGFR2 Ser252Trp, dan atrofi diskus optikus lebih umum terjadi pada pasien dengan mutasi FGFR2 Pro253Arg. Pasien dengan mutasi FGR2 Ser252Trp memiliki prevalensi gangguan penglihatan yang jauh lebih tinggi dibandingkan dengan pasien dengan mutasi FGFR2 Pro253Arg.
Gejala Sindrom Aper
Beberapa manifestasi penyakit ini sudah terlihat jelas sejak bayi lahir, karena penyakit ini berkembang di dalam rahim ibu. Di antara gejala utama sindrom ini:
- Tengkorak mengalami deformasi - tengkorak melebar tinggi, sehingga tampak seperti "menara"; selain itu, mata melebar dan sedikit menonjol (karena rongga mata mengecil); hidung melebar dan gigitan tidak normal terbentuk (gigi atas menonjol berlebihan);
- Jari-jari anggota badan menyatu sempurna (terutama jari manis, begitu pula jari tengah dan jari telunjuk) dan tampak seperti selaput kulit atau penyatuan tulang yang lengkap; selain itu, jari-jari tambahan mungkin tumbuh;
- Keterbelakangan mental (tidak terjadi pada semua orang);
- Saraf optik mengalami atrofi, yang mengakibatkan penurunan ketajaman penglihatan (pada beberapa kasus, terjadi kehilangan penglihatan total);
- Tekanan intrakranial yang meningkat, yang terjadi akibat penutupan dini sutura kranial, bermanifestasi dalam bentuk sakit kepala dan muntah disertai mual;
- Karena rahang atas masih belum berkembang, timbul masalah pernafasan;
- Sindrom apnea tidur merupakan kejadian umum.
- Manifestasi emosional – agresi, kurangnya pengendalian diri, lekas marah yang hebat.
Komplikasi dan konsekuensinya
Sebagian besar pasien mengalami beberapa derajat penyumbatan saluran napas bagian atas pada masa bayi. Saluran napas bagian atas tidak terlindungi dengan baik karena ukuran nasofaring dan koana yang mengecil, dan saluran napas bagian bawah dapat menyebabkan kematian dini karena kelainan tulang rawan trakea.
Diagnostik Sindrom Aper
Untuk membuat diagnosis, langkah-langkah berikut diperlukan:
- Dokter harus menganalisis keluhan pasien, serta riwayat medisnya. Perlu diketahui apakah ada kasus patologi serupa dalam keluarga;
- Pemeriksaan neurologis untuk menilai bentuk tengkorak dan perkembangan intelektual pasien (kuesioner khusus serta wawancara);
- Pemeriksaan fundus untuk mendeteksi adanya gejala peningkatan tekanan intrakranial (pembengkakan diskus optikus, serta pengaburan tepinya);
- Untuk menilai kondisi tengkorak, dilakukan rontgen;
- Pencitraan komputer dan resonansi magnetik kepala untuk memeriksa struktur otak dan tengkorak lapis demi lapis, untuk menentukan adanya gejala fusi prematur sutura kranial, dan sebagai tambahan, hidrosefalus (akibat peningkatan tekanan intrakranial, terakumulasi cairan serebrospinal berlebih (ini adalah cairan serebrospinal yang meningkatkan proses metabolisme, serta nutrisi otak));
- Rontgen kaki dan tangan untuk menentukan penyebab menyatunya jari-jari (ini penting untuk merencanakan intervensi bedah selanjutnya);
- Konsultasi dengan ahli genetika medis dan ahli bedah saraf mungkin akan diresepkan.
Tes
Analisis genetik terhadap mutasi umum yang terjadi pada gen tipe FGFR2 sedang dilakukan.
Perbedaan diagnosa
Sindrom ini harus dibedakan dari patologi genetik lain yang menunjukkan adanya craniosynostosis. Penyakit-penyakit ini adalah sindrom Pfeiffer, Crouzon, Saethre-Chotzen, dan Carpenter. Metode pengujian genetik molekular digunakan untuk menyingkirkan anomali ini.
Siapa yang harus dihubungi?
Pengobatan Sindrom Aper
Pembedahan dianggap satu-satunya pengobatan yang efektif untuk sindrom Apert – pembedahan membantu memperbaiki cacat fisik individu dan juga memperbaiki keterbelakangan mental.
Selama prosedur ini, sutura koronal ditutup untuk mencegah kemungkinan cedera otak. Teknik yang paling umum adalah distraksi kraniofasial, yang melibatkan traksi tengkorak bertahap. Bedah ortodontik dan/atau ortognatik dilakukan untuk menghilangkan cacat wajah individual.
Selain itu, pasien menjalani koreksi bedah fusi jari.
Referensi
Genetika Medis. Panduan Nasional. Diedit oleh Ginter EK, Puzyrev VP GEOTAR-Media. 2022