Ahli medis artikel

Publikasi baru
Mucopolysaccharidosis tipe 3
Terakhir ditinjau: 04.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
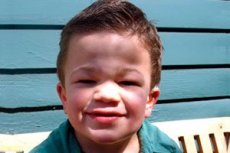
Mukopolisakaridosis, tipe III (sinonim: sindrom Sanfilippo, defisiensi lisosomal aN-asetilglukosaminidase - mukopolisakaridosis III A, defisiensi asetil-CoA-a-glukosaminid-N-asetiltransferase - mukopolisakaridosis III B, N-asetilglukosamin-6-sulfatase - mukopolisakaridosis III C, defisiensi sulfamidase - mukopolisakaridosis III D).
 [
[ Patogenesis
Penyakit ini disebabkan oleh mutasi pada empat gen yang berbeda: lisosomal aN-asetilglukosaminidase (mukopolisakarida III A), asetil-CoA-a-glukosaminid-N-asetiltransferase (mukopolisakarida III B), lisosomal N-asetilglukosamin-6-sulfatase (mukopolisakarida III C), dan sulfamidase (mukopolisakarida III D). Semua enzim terlibat dalam metabolisme heparan sulfat.
Gen heparan-N-sulfatase - SGSH - terletak pada lengan panjang kromosom 17 - 17q25.3. Sebanyak 75,3% mutasi yang diketahui saat ini pada gen SGSH adalah mutasi titik. Mutasi yang sering terjadi yang menjadi ciri populasi Eropa telah dijelaskan - R74C (56% di Polandia dan 21% di Jerman) dan R245H (56% di Belanda).
Frekuensi mutasi R74C adalah 47,5%, mutasi R245H adalah 7,5%. Dua mutasi lain yang dijelaskan, delll35G dan N389S, bersama-sama mencakup 21,7% alel mutan.
Gen untuk aN-acetyl-glucosaminidase (NAGLU) terletak pada lengan panjang kromosom 17 - 17q21. 69% mutasi yang ditemukan pada gen NAGLU adalah mutasi missense dan nonsense, 26,3% adalah delesi dan insersi kecil. Gen untuk acetyl-CoA-cc-glucosaminid-N-acetyltransferase (HGSNAT) terletak pada lengan pendek kromosom 8 - 8p11.1. Gen tersebut baru dikarakterisasi pada tahun 2006 dan hingga saat ini hanya beberapa mutasi yang ditemukan di dalamnya.
Gen N-asetil-glukosamin-6-sulfatase - GNS - terletak pada lengan panjang kromosom 12 - 12ql4. Ada 12 pasien dengan mukopolisakaridosis IIID yang terdaftar di dunia. Empat mutasi pada gen GNS telah dijelaskan.
Pada semua subtipe mukopolisakaridosis III, terdapat gangguan pada degradasi heparan sulfat, yang merupakan bagian dari struktur membran sel, termasuk membran neuronal, yang berkorelasi dengan proses neurodegeneratif berat yang disebabkan oleh atrofi kortikal. Diare kronis dijelaskan oleh keterlibatan sistem saraf otonom dalam proses patologis bersama dengan disfungsi mukosa usus. Gangguan pendengaran sensorineural mungkin disebabkan oleh tiga penyebab: otitis yang sering, deformasi tulang pendengaran, dan anomali telinga bagian dalam. Kekakuan sendi merupakan hasil dari deformasi metafisis, penebalan kapsul sendi merupakan akibat sekunder dari pengendapan glikosaminoglikan dan fibrosis. Perbedaan intra-sindromik dalam tingkat keparahan penyakit semata-mata disebabkan oleh aktivitas fungsional residual dari enzim mutan: semakin tinggi, semakin ringan penyakitnya.
Gejala mukopolisakaridosis tipe 3
Polimorfisme klinis pada sindrom Sanfilippo kurang menonjol dibandingkan dengan jenis mukopolisakaridosis lainnya. Perkembangan penyakit yang lambat, gangguan neurologis yang parah dengan gejala ringan dari organ dalam dan sistem lainnya merupakan ciri khasnya.
Gejala pertama penyakit ini biasanya muncul antara usia 2 dan 6 tahun pada anak-anak yang sebelumnya memiliki perkembangan normal. Gejala yang nyata meliputi kemunduran perkembangan psikomotorik dan bicara, gangguan kejiwaan berupa sindrom hiperaktif, perilaku autis atau agresif, gangguan tidur; anak-anak menjadi ceroboh dan kurang perhatian.
Gejala umum lainnya adalah hirsutisme, rambut kasar, hepatosplenomegali sedang, deformitas valgus pada tungkai, dan leher pendek. Perkembangan fitur wajah kasar seperti gargoyilisme dan deformitas rangka seperti disostosis multipel diekspresikan dengan lemah pada mukopolisakaridosis III dibandingkan dengan jenis mukopolisakaridosis lain yang ditandai dengan fenotipe Hurler. Tinggi badan, sebagai aturan, sesuai dengan usia, dan kekakuan sendi jarang menyebabkan disfungsi. Kebanyakan pasien sering mengalami osteoporosis dan osteomalasia. Gangguan rangka sekunder - risiko tinggi patah tulang patologis. Gangguan psikoneurologis yang parah paling sering diamati pada tahun ke-6-10 kehidupan, yang menyebabkan maladjustment sosial yang nyata. Kehilangan pendengaran sensorineural progresif melekat pada semua pasien dengan bentuk penyakit yang parah dan sedang. Kejang diamati pada hampir semua pasien saat penyakit berkembang.
Penyakit ini berkembang dengan cepat, dan sebagian besar pasien tidak bertahan hidup hingga usia 20 tahun. Mukopolisakaridosis IIIA dianggap sebagai jenis sindrom ini yang paling umum dan parah.
Diagnostik mukopolisakaridosis tipe 3
Diagnosis mukopolisakaridosis III dipastikan dengan menentukan kadar ekskresi glikosaminoglikan urin dan mengukur aktivitas enzim. Dalam kasus mukopolisakaridosis III, total ekskresi glikosaminoglikan dalam urin meningkat dan terjadi hiperekskresi heparan sulfat. Aktivitas enzim lisosom yang sesuai dengan subtipe tertentu mukopolisakaridosis III diukur dalam leukosit atau kultur fibroblas kulit menggunakan substrat fluorogenik buatan.
Diagnostik prenatal dapat dilakukan dengan mengukur aktivitas enzim dalam biopsi vili korionik pada usia kehamilan 9-11 minggu dan/atau menentukan spektrum glikosaminoglikan dalam cairan ketuban pada usia kehamilan 20-22 minggu. Bagi keluarga dengan genotipe yang diketahui, diagnostik DNA dapat dilakukan pada awal kehamilan.
Tes apa yang dibutuhkan?
Perbedaan diagnosa
Diagnostik diferensial dilakukan baik dalam kelompok mukopolisakaridosis dan dengan penyakit penyimpanan lisosomal lainnya: mukolipidosis, galaktosialidosis, sialidosis, mannosidosis, fukosidosis, gangliosidosis GM1.
Siapa yang harus dihubungi?
Pengobatan mukopolisakaridosis tipe 3
Hingga saat ini, metode terapi yang efektif untuk mukopolisakaridosis III belum dikembangkan. Terapi simptomatik diindikasikan.
Использованная литература