Ahli medis artikel

Publikasi baru
Sindrom Angelman pada anak-anak dan orang dewasa
Terakhir ditinjau: 04.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.

Ada sejumlah penyakit yang ungkapan seperti "jaga diri baik-baik dan kamu tidak akan sakit" terdengar konyol. Ini adalah patologi di mana beberapa kelainan mental dan fisik sudah ada dalam tubuh anak bahkan sebelum lahir, tetapi orang tua tidak dapat disalahkan untuk ini. Penyakit-penyakit tersebut disebabkan oleh mutasi atau kelainan pada set kromosom dan disebut kromosom atau genetik. Sindrom Angelman, sindrom Down, sindrom Patau, sindrom Edwards, sindrom Turner, sindrom Prader-Willi - ini hanya sebagian dari penyakit genetik dari daftar yang cukup bagus.
Sindrom Pria Bahagia
Kali ini kita akan membahas tentang patologi yang dinamai menurut dokter anak Inggris Harry Angelman, yang pertama kali mengangkat masalah ini pada tahun 1965, setelah menemui tiga anak yang tidak biasa dalam praktiknya sehari sebelumnya, yang disatukan oleh gejala-gejala aneh yang umum. Dokter tersebut menyebut anak-anak ini sebagai anak boneka dan menulis sebuah artikel tentang mereka, yang awalnya disebut "Anak-anak boneka". Artikel itu sendiri dan judulnya ditulis berdasarkan kesan sebuah lukisan yang terlihat di salah satu museum di Verona. Lukisan itu menggambarkan seorang anak laki-laki yang sedang tertawa, dan disebut "Anak Boneka". Keterkaitan anak yang digambarkan dalam lukisan itu dengan tiga anak yang pernah ditemui Angelman dalam praktiknya mendorong dokter anak tersebut untuk menggabungkan anak-anak tersebut ke dalam satu kelompok karena penyakit yang mereka derita.
Tidak mengherankan jika anak-anak yang disebutkan dalam artikel tersebut tidak diperhatikan oleh dokter lain. Lagi pula, sekilas tampak bahwa mereka memiliki penyakit yang sama sekali berbeda, gambaran klinis umum penyakit tersebut dalam 3 kasus berbeda sangat berbeda. Mungkin patologi kromosom "baru" akan menarik minat ilmuwan lain, tetapi pada saat itu genetika belum cukup berkembang untuk mengonfirmasi hipotesis dokter Inggris tersebut. Oleh karena itu, setelah ada minat tertentu terhadapnya, artikel tersebut dibuang ke rak belakang untuk waktu yang lama.
Penyebutan sindrom Angelman berikutnya, yang sekarang menjadi judul artikel dokter anak Inggris G. Angelman, muncul pada awal tahun 80-an abad ke-20. Baru pada tahun 1987 ditemukan alasan mengapa sebagian kecil anak terlahir dengan kelainan sedemikian rupa sehingga dari luar mereka tampak selalu tersenyum dan bahagia. Padahal, ini sama sekali tidak benar, dan senyum hanyalah seringai, yang di baliknya tersembunyi jiwa manusia yang tidak bahagia dan rasa sakit orang tua.
Epidemiologi
Menurut statistik, mutasi kromosom pada anak dapat berkembang baik dengan latar belakang mutasi serupa pada orang tua maupun tanpa adanya mutasi serupa. Tidak ada sifat keturunan yang jelas dari sindrom Angelman (AS), tetapi kemungkinan berkembangnya patologi pada orang tua dengan mutasi kromosom cukup tinggi.
Yang juga menarik, jika sebuah keluarga sudah memiliki anak dengan AS, ada satu persen kemungkinan memiliki anak kedua dengan gangguan yang sama, bahkan jika orang tuanya sehat.
Hingga saat ini belum ada statistik pasti mengenai jumlah pasien dengan sindrom Angelman. Mungkin penyebabnya adalah berbagai gejala, yang dapat terjadi dalam komposisi tertentu atau tidak terjadi sama sekali dalam jangka waktu yang lama. Diasumsikan bahwa prevalensi penyakit ini adalah: 1 anak per 20.000 bayi baru lahir. Namun, angka ini sangat mendekati.
Penyebab Sindrom Angelman
Sindrom Angelman adalah nama medis untuk kelainan kromosom, tetapi bukan satu-satunya. Orang-orang menyebut penyakit ini sindrom anak boneka, sindrom boneka bahagia, sindrom Petrushka, dan sindrom boneka tertawa. Orang-orang memberikan berbagai macam nama (kadang-kadang bahkan menyinggung pasien itu sendiri dan orang tua mereka), tetapi penyakit adalah penyakit, tidak peduli seberapa lucu kelihatannya dan tidak peduli apa pun alasannya.
Dan alasan perkembangan sindrom Angelman, seperti banyak patologi genetik lainnya, dalam semua kasus adalah gangguan pada struktur salah satu kromosom atau set kromosom secara keseluruhan. Namun dalam kasus kami, seluruh masalahnya terletak pada kromosom 15, yang diturunkan dari ibu. Artinya, kromosom paternal dalam kasus ini tidak memiliki penyimpangan, tetapi kromosom betina mengalami mutasi tertentu.
Berdasarkan jenis kelainan kromosom, sindrom Angelman diklasifikasikan sebagai mutasi kromosom. Mutasi tersebut dianggap sebagai:
- Penghapusan (ketiadaan bagian kromosom yang mengandung sekumpulan gen tertentu; jika salah satu gen hilang, kita berbicara tentang mikrodelesi), yang merupakan hasil dari dua kali kerusakan dan satu kali penyatuan kembali, ketika satu bagian kromosom asli hilang.
- Duplikasi (adanya bagian tambahan pada kromosom yang merupakan salinan dari bagian yang sudah ada), yang dalam banyak kasus menyebabkan kematian seseorang, dan yang lebih jarang menyebabkan kemandulan.
- Inversi (pembalikan salah satu bagian kromosom sebesar 180 derajat, yaitu ke arah yang berlawanan, kemudian gen-gen di dalamnya tersusun dalam urutan yang berlawanan), yakni ketika ujung-ujung kromosom yang putus disambung dengan urutan yang berbeda dari semula.
- Insersi (jika bagian materi genetik dalam kromosom tidak pada tempatnya),
- translokasi (jika bagian tertentu dari kromosom melekat pada kromosom lain; mutasi semacam itu dapat terjadi secara timbal balik tanpa kehilangan bagian).
Menerima kromosom yang bermutasi dari ibu yang tidak curiga, anak tersebut ditakdirkan untuk lahir dengan kelainan. Penyebab paling umum dari sindrom Angelman masih dianggap sebagai penghapusan kromosom ke-15 ibu, ketika sebagian kecil hilang. Mutasi yang kurang umum dalam sindrom "boneka tertawa" dianggap sebagai:
- translokasi,
- disomi unipaternal (jika anak menerima sepasang kromosom dari ayah, kromosom ibu tidak ada),
- mutasi gen dalam DNA, yang merupakan bahan pembangun utama (genetik) dan petunjuk untuk penggunaan yang benar (khususnya, mutasi gen ube3a dalam kromosom ibu).
Kehadiran salah satu mutasi ini pada orang tua merupakan faktor risiko perkembangan sindrom Angelman pada anak-anak. Namun, tidak hanya mutasi kromosom, tetapi juga mutasi genomik (yang dikaitkan dengan perubahan kuantitatif pada set kromosom dan lebih umum daripada mutasi kromosom) dapat memicu perkembangan penyakit pada anak. Mutasi genomik yang umum termasuk trisomi kromosom (jika set kromosom seseorang memiliki lebih dari 46 kromosom).
Agar suatu patologi muncul pada seorang anak, sama sekali tidak perlu orangtuanya memiliki kelainan kromosom. Namun, ada persentase tertentu dari pasien yang penyakitnya bersifat turun-temurun.
Patogenesis
Mari kita gali lebih dalam tentang biologi, atau lebih tepatnya genetika. Informasi genetik setiap individu organisme manusia terkandung dalam 23 pasang kromosom. Satu kromosom dari sepasang kromosom diwariskan kepada anak dari ayah, yang lain dari ibu. Semua pasang kromosom berbeda dalam bentuk dan ukuran dan membawa informasi tertentu. Dengan demikian, pasangan kromosom ke-23 (kromosom X dan Y) bertanggung jawab atas pembentukan karakteristik seksual bayi (XX - perempuan, XY - laki-laki, sedangkan kromosom Y hanya dapat diterima oleh anak dari ayah).
Idealnya, seorang anak menerima 46 kromosom dari orang tuanya, yang membentuk karakteristik genetiknya, yang menentukannya sebagai seorang individu. Jumlah kromosom yang lebih banyak disebut trisomi dan dianggap sebagai penyimpangan dari norma. Misalnya, keberadaan kromosom 47 dalam set kromosom (kariotipe, yang menentukan spesies dan karakteristik individu) menyebabkan terjadinya sindrom Down.
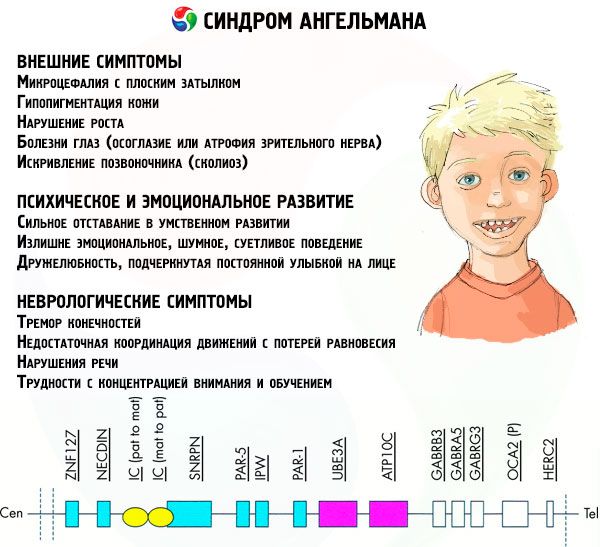
Jika kromosom diwarnai dengan pewarna khusus, maka di bawah mikroskop Anda dapat melihat garis-garis dengan corak yang berbeda di sepanjang masing-masing kromosom. Di dalam setiap garis terdapat sejumlah besar gen. Semua garis ini diberi nomor oleh para ilmuwan dan memiliki lokasi yang tetap. Tidak adanya salah satu garis dianggap sebagai penyimpangan dari norma. Pada sindrom Angelman, seseorang dapat sangat sering mengamati tidak adanya segmen kromosom ibu dalam interval q11-q13, yang terletak di lengan panjang, jumlah basa DNA di mana hanya sekitar 4 juta.
Komponen utama kromosom dianggap sebagai molekul DNA yang sangat panjang yang mengandung ribuan gen dan puluhan hingga ratusan juta basa nitrogen. Jadi, kromosom 15, yang bertanggung jawab atas perkembangan sindrom Angelman dan beberapa lainnya, mengandung 1.200 gen dan sekitar 100 juta basa. Setiap gangguan dalam struktur molekul DNA tentu akan memengaruhi penampilan dan perkembangan anak di masa depan.
Informasi genetik yang terkandung dalam gen diubah menjadi protein atau RNA. Proses ini disebut ekspresi gen. Dengan cara ini, informasi genetik yang diterima dari orang tua menerima bentuk dan isi, yang diwujudkan dalam pewaris perempuan atau laki-laki mereka yang unik.
Ada sejumlah patologi dengan jenis pewarisan non-klasik, termasuk sindrom Angelman, di mana gen yang diterima dari orang tua sebagai bagian dari kromosom berpasangan memiliki jejak unik orang tua dan memanifestasikan dirinya dalam cara yang berbeda.
Jadi, sindrom Angelman adalah contoh mencolok dari genomic imprinting, di mana ekspresi gen dalam tubuh anak secara langsung bergantung pada orang tua mana alel tersebut diterima (berbagai bentuk gen yang sama, yang diterima dari ayah dan ibu, terletak pada bagian identik dari kromosom berpasangan). Artinya, hanya anomali pada kromosom ibu yang menyebabkan perkembangan sindrom, sementara mutasi dan kelainan struktural pada kromosom ayah menyebabkan patologi yang sama sekali berbeda.

Pada patologi ini, terdapat kekurangan gen tertentu pada kromosom ibu atau kehilangan/penurunan aktivitas gen individual (pada sebagian besar kasus, gen ube3a, yang terlibat dalam metabolisme ubiquitin, protein yang mengatur degradasi protein lain). Akibatnya, anak didiagnosis dengan kelainan perkembangan mental dan kelainan bentuk fisik.
Gejala Sindrom Angelman
Gejala sindrom Angelman memengaruhi berbagai aspek kehidupan dan perkembangan anak: fisik, neurologis, dan mental. Berdasarkan hal ini, 3 kelompok gejala dapat diidentifikasi yang menunjukkan perkembangan patologi ini.
- Gejala eksternal atau fisik:
- kepala yang ukurannya sangat kecil dibandingkan dengan ukuran tubuh dan anggota tubuh yang normal,
- mulut terlalu lebar,
- hampir selalu ada senyum di wajah (dengan mulut terbuka),
- gigi jarang,
- bibir atas sempit,
- sering menjulurkan lidah lebar,
- rahang bawah menonjol,
- dagu runcing,
- kulit sangat terang, seringkali berambut (albinisme, terkait dengan fakta bahwa tubuh tidak menghasilkan pigmen melanin),
- bintik hitam pada kulit putih (hipopigmentasi karena produksi melanin yang tidak mencukupi)
- gejala fisik atau eksternal: penyakit mata seperti strabismus atau atrofi saraf optik,
- kelengkungan tulang belakang (skoliosis),
- kaki kaku (ketika berjalan, seseorang tidak menekuk kakinya di lutut karena mobilitas sendi yang rendah, maka dibandingkan dengan gaya berjalan boneka).
- Gejala yang berhubungan dengan perkembangan mental dan emosional:
- keterbelakangan mental yang parah,
- perilaku yang terlalu emosional, berisik, cerewet,
- sering bertepuk tangan,
- mengungkapkan keramahan, ditekankan dengan senyum yang terus-menerus di wajah,
- sering tertawa tanpa alasan.
- Gejala neurologis:
- getaran anggota badan,
- koordinasi gerakan yang tidak memadai dengan hilangnya keseimbangan,
- penurunan tonus otot,
- berbagai gangguan tidur,
- sering mengalami kejang histeris di masa kecil,
- gangguan bicara (anak mulai berbicara terlambat, memiliki keterampilan komunikasi yang buruk dan bicara tidak jelas),
- hiperaktif dengan latar belakang peningkatan rangsangan,
- kesulitan berkonsentrasi dan belajar.
Namun, ini adalah gambaran umum penyakit tersebut. Faktanya, gambaran klinis sindrom Angelman sangat bergantung pada tahap perkembangan penyakit dan jenis mutasi kromosom yang menyebabkan patologi tersebut. Ini berarti bahwa gejala penyakit dapat berbeda secara signifikan pada pasien yang berbeda, yang untuk waktu yang lama tidak memungkinkan kita untuk membedakan patologi tersebut dari yang lain dengan gambaran klinis yang serupa.
Di antara semua gejala yang ada, kita dapat menyoroti gejala-gejala yang menjadi ciri khas semua pasien tanpa kecuali:
- keterbelakangan mental yang parah,
- perilaku yang tidak pantas (tertawa terbahak-bahak, peningkatan rangsangan, konsentrasi buruk, keadaan euforia),
- kurangnya perkembangan keterampilan motorik,
- koordinasi gerakan yang buruk, ataksia gaya berjalan (kecepatan tidak merata, bergoyang dari sisi ke sisi, dll.), tremor pada anggota badan.
- gangguan perkembangan bicara dengan dominasi cara komunikasi non-verbal.
Di antara gejala-gejala yang dialami oleh sebagian besar pasien, berikut ini dapat dibedakan:
- ketidakseimbangan antara kepala dan tubuh yang disebabkan oleh keterlambatan perkembangan fisik,
- pada banyak pasien bentuk tengkoraknya sedemikian rupa sehingga ukuran otaknya tetap lebih kecil dibandingkan orang sehat (mikrosefali),
- kejang epilepsi sebelum usia 3 tahun dengan penurunan kekuatan dan frekuensi secara progresif pada usia yang lebih tua,
- distorsi parameter EEG (fluktuasi dan amplitudo tinggi gelombang frekuensi rendah).
Gejala-gejala ini cukup umum, namun 20% pasien dengan sindrom Angelman tidak memilikinya.
Bahkan lebih jarang lagi, adalah mungkin untuk mendiagnosis manifestasi penyakit seperti:
- strabismus parah atau ringan,
- kontrol gerakan lidah yang buruk, sehingga pasien sering menjulurkan lidah tanpa alasan,
- kesulitan menelan dan menghisap, terutama pada anak kecil,
- gangguan pigmentasi kulit dan mata,
- lengan diangkat atau ditekuk saat berjalan,
- hiperrefleksia,
- gangguan tidur, terutama pada masa kanak-kanak,
- sering mengeluarkan air liur,
- haus yang tak terpuaskan,
- gerakan mengunyah yang terlalu aktif,
- hipersensitivitas terhadap panas,
- bagian belakang kepala datar,
- rahang bawah menonjol,
- telapak tangan halus.
Persentase pasien yang cukup besar mengalami masalah buang air kecil yang tidak dapat mereka kendalikan dengan baik, gangguan keterampilan motorik halus yang menyebabkan kesulitan dalam perawatan diri dan belajar, serta kelebihan berat badan. Hampir semua pasien mengalami pubertas lebih lambat daripada rekan-rekan mereka yang sehat.
Anak-anak dengan sindrom Angelman memahami dan menghayati pembicaraan lisan dengan baik, tetapi tidak mau berpartisipasi dalam percakapan, sehingga ucapan mereka terbatas pada beberapa lusin kata yang diperlukan dalam kehidupan sehari-hari. Namun, di masa dewasa, pasien tersebut terlihat lebih muda daripada teman sebayanya yang tidak memiliki kelainan genetik.
Banyak gejala sindrom Angelman yang tidak permanen, sehingga gambaran klinis penyakit ini berubah secara signifikan seiring bertambahnya usia. Kejang dan serangan epilepsi menjadi lebih jarang atau hilang sama sekali, pasien menjadi kurang bersemangat, dan tidur membaik.
Komplikasi dan konsekuensinya
Sindrom Angelman adalah kelainan kromosom yang parah dan hampir tidak dapat disembuhkan yang menghilangkan kesempatan pasien untuk menjalani kehidupan normal. Seperti apa kehidupan anak dengan AS akan sangat bergantung pada jenis kelainan kromosom.
Duplikasi segmen kromosom tidak sesuai dengan kehidupan dalam banyak kasus. Dan bahkan jika pasien tersebut tidak meninggal saat masih bayi dan mencapai pubertas, mereka tidak memiliki peluang untuk memiliki anak.
Hilangnya atau tidak adanya sebagian gen yang paling sering terjadi pada sindrom Angelman merupakan hambatan bagi anak untuk belajar berjalan dan berbicara. Anak-anak tersebut memiliki bentuk keterbelakangan mental yang lebih parah, dan kejang epilepsi lebih sering terjadi, dan intensitasnya jauh lebih besar daripada pada pasien dengan kelainan kromosom lainnya.
Jika hanya terjadi mutasi pada satu gen, dengan perhatian dan pendekatan yang tepat, anak dapat diajarkan dasar-dasar perawatan diri, komunikasi, dan interaksi dalam kelompok, meskipun ia masih akan tertinggal dari teman-temannya dalam perkembangan.
Bagi anak-anak penderita sindrom Angelman, yang pada dasarnya baik hati, yang terpenting adalah kasih sayang dan perhatian dari orang tua. Hanya dalam kasus ini pendidikan anak akan membuahkan hasil, meskipun kecil. Tentu saja, penderita AS tidak akan dapat belajar di sekolah biasa. Mereka membutuhkan kelas khusus di mana anak-anak akan terlebih dahulu diajarkan untuk berkonsentrasi, dan kemudian secara bertahap akan diberikan dasar-dasar pengetahuan sekolah.
Diagnostik Sindrom Angelman
Sindrom Angelman merupakan kelainan perkembangan bawaan. Namun, karena keadaan tertentu, sering kali tidak mungkin untuk mendiagnosisnya pada masa bayi dan anak usia dini. Hal ini disebabkan oleh ketidakspesifikan dan ekspresi gejala yang lemah pada bayi dan anak di bawah usia 3 tahun. Dan prevalensi penyakit ini di negara kita tidak begitu besar sehingga dokter telah belajar untuk mengenalinya di antara rekan-rekannya.
Sindrom Angelman pada bayi dapat bermanifestasi sebagai penurunan tonus otot, yang bermanifestasi dalam bentuk masalah makan (kelemahan refleks mengisap dan menelan), dan kemudian kesulitan belajar berjalan (anak-anak seperti itu mulai berjalan jauh lebih lambat). Gejala-gejala ini adalah tanda-tanda pertama kelainan perkembangan pada bayi, yang mungkin terkait dengan kelainan kromosom. Hanya analisis genetik yang dapat mengonfirmasi asumsi ini.
Perhatian khusus diberikan kepada anak-anak yang orang tuanya memiliki berbagai kelainan genomik atau kromosom. Bagaimanapun, penyakit ini mungkin tidak menampakkan dirinya pada awalnya, dan jika patologi terdeteksi tepat waktu, dengan mulai bekerja secara intensif dengan anak, adalah mungkin untuk mencapai keberhasilan yang jauh lebih besar dalam pembelajaran, memperlambat perkembangan penyakit.
Jika orang tua memiliki berbagai kelainan kromosom, analisis genetik dilakukan bahkan sebelum bayi lahir, karena SA merupakan salah satu patologi yang dapat dideteksi pada tahap embrio.
Pengumpulan bahan untuk penelitian genetik dapat dilakukan dengan dua cara:
- invasif (dengan persentase risiko tertentu, karena perlu menembus rahim untuk mengambil sampel cairan ketuban),
- non-invasif (analisis DNA bayi dari darah ibu).
Penelitian berikut kemudian dilakukan:
- hibridisasi fluoresensi in situ (metode FISH) – pengikatan probe DNA yang diberi label dengan pewarna khusus ke DNA yang sedang dipelajari, diikuti dengan pemeriksaan di bawah mikroskop.
- analisis mutasi pada gen ube3a dan gen yang dicetak,
- Analisis metilasi DNA menggunakan metode khusus yang digunakan dalam genetika.
Tes genetik memberikan informasi yang cukup akurat dalam kasus kelainan kromosom, yang berarti bahwa calon orang tua mengetahui terlebih dahulu apa yang harus dipersiapkan. Namun, ada pengecualian. Pada sekelompok pasien tertentu, dengan adanya semua gejala yang menunjukkan patologi, hasil tes tetap normal. Artinya, patologi hanya dapat diidentifikasi dengan mengamati anak secara cermat sejak usia dini: bagaimana ia makan, kapan ia mulai berjalan dan berbicara, apakah ia menekuk kakinya saat berjalan, dll.
Selain metode FISH, di antara metode diagnostik instrumental untuk sindrom Angelman, seseorang dapat memilih tomografi (CT atau MRI), yang membantu menentukan kondisi dan ukuran otak, dan elektroensefalogram (EEG), yang menunjukkan cara kerja masing-masing bagian otak.
Dokter biasanya membuat diagnosis akhir pada usia 3-7 tahun, ketika pasien sudah memiliki sebagian besar gejala dan dinamika perkembangan penyakit terlihat.
Tes apa yang dibutuhkan?
Perbedaan diagnosa
Sindrom Angelman adalah patologi genetik yang hampir tidak memiliki manifestasi spesifik. Sebagian besar gejala dapat mengindikasikan AS dan patologi genetik lainnya.
Diagnosis banding sindrom Angelman dilakukan dengan patologi berikut:
- Sindrom Pitt-Hopkins (pasien ditandai dengan keterbelakangan mental, karakter ceria, suka tersenyum, memiliki mulut yang agak besar dan lebar, mikrosefali dicatat). Perbedaannya adalah serangan hiperventilasi dan menahan napas dalam keadaan terjaga.
- Sindrom Christianson (penderita merupakan penyandang retardasi mental, berwatak ceria, tidak mampu berbicara, ditandai dengan mikrosefali, ataksia, kejang, gerakan otot tak terkendali).
- Sindrom Mowat-Wilson (gejala: keterbelakangan mental, kejang epilepsi, dagu lancip, mulut terbuka, ekspresi wajah gembira, mikrosefali). Ciri khas: jarak antar mata jauh, mata sipit ke dalam, ujung hidung membulat, daun telinga terbalik.
- Sindrom Kabuki (ditandai dengan retardasi mental ringan hingga sedang, masalah bicara dan motorik, kelemahan otot, kejang epilepsi, mikrosefali, interval panjang antara gatal, dan gangguan koordinasi). Ditandai dengan alis melengkung, bagian lateral kelopak mata bawah terbalik, mata lebar, fisura palpebra panjang dengan bulu mata panjang dan tebal.
- Sindrom Rett (diferensiasi dari AS pada wanita). Gejala: perkembangan bicara tertunda, kejang, mikrosefali. Perbedaannya adalah tidak ada ekspresi bahagia di wajah, ada serangan apnea dan apraksia, yang berkembang seiring waktu.
- Sindrom retardasi mental resesif autosomal 38 (gejala: retardasi mental yang nyata disertai keterlambatan dalam keterampilan motorik dan bicara, kelemahan otot, masalah makan pada masa bayi, impulsivitas). Ciri khasnya adalah warna biru pada iris mata.
- Sindrom duplikasi gen MECP 2 (diferensiasi dari SA pada pria). Gejala: retardasi mental berat, kelemahan otot sejak masa kanak-kanak, masalah bicara atau kurang bicara, epilepsi. Perbedaan: miopati progresif, infeksi yang terus-menerus berulang.
- Sindrom Kleefstra (gejala: masalah bicara dan berpikir, kelemahan otot, gangguan tidur, kurang perhatian, mulut terbuka, hiperaktif, kejang, ataksia, gangguan keseimbangan). Ciri khas: wajah datar, hidung pesek pendek, mata cekung, bibir bawah besar terbalik, ledakan agresif.
- Sindrom Smith-Magenis (ditandai dengan kejang, masalah tidur, gangguan perkembangan intelektual dan motorik). Ciri khasnya meliputi wajah lebar dan datar serta dahi menonjol.
- Sindrom Koolen-de Vries (keterbelakangan mental ringan hingga sedang, kelemahan otot, kejang, keramahan). Ciri-ciri yang membedakan: wajah panjang dengan dahi tinggi, telinga menonjol, mata sipit, mobilitas sendi tinggi, cacat jantung bawaan.
- Sindrom Phelan-McDermid (gejala: keterbelakangan mental, gangguan bicara atau tidak bisa bicara). Ciri-ciri: tangan besar dengan otot yang berkembang, kelemahan otot sejak lahir, keringat yang lemah.
Patologi seperti defisiensi adenil suksinat, sindrom retardasi mental resesif autosomal 1, sindrom duplikasi kromosom 2q23.1, sindrom haploinsufisiensi gen FOXG1, STXBP1 atau MEF2C dan beberapa lainnya dapat “membanggakan” gejala yang mirip dengan sindrom Angelman.
Tugas dokter adalah membuat diagnosis yang akurat, membedakan sindrom Angelman dari patologi dengan gejala serupa, dan meresepkan pengobatan efektif yang relevan dengan stadium penyakit yang didiagnosis.
Siapa yang harus dihubungi?
Pengobatan Sindrom Angelman
Sindrom Angelman merupakan salah satu patologi yang masih dalam tahap pencarian pengobatan yang efektif oleh dunia kedokteran. Pengobatan etiologi penyakit ini masih dalam tahap pengembangan berbagai metode dan cara, yang banyak di antaranya belum diuji pada manusia. Ini berarti bahwa untuk saat ini dokter harus membatasi diri pada terapi simptomatik, yang membantu meringankan situasi yang tidak mengenakkan bagi anak-anak dan orang dewasa dengan sindrom marionette, yang menderita kejang epilepsi, air liur berlebihan, hipotensi, dan gangguan tidur.
Dengan demikian, frekuensi dan kekuatan kejang epilepsi dapat dikurangi dengan bantuan obat antikonvulsan yang dipilih dengan tepat. Namun, kesulitannya adalah bahwa kejang pada pasien SA berbeda dari kejang epilepsi biasa karena kejang tersebut ditandai oleh beberapa jenis kejang, yang berarti bahwa kondisi tersebut dapat diringankan dengan pemberian beberapa obat sekaligus.
Antikonvulsan yang paling populer digunakan untuk mengobati sindrom Angelman adalah: asam valproat, topiramate, lamotrigin, levetiracetam, klonazepam, dan obat-obatan yang dibuat berdasarkan obat-obatan tersebut. Yang kurang umum digunakan adalah obat-obatan yang dibuat berdasarkan karmazepin, fenitoin, fenobarbital, etosuksimida, karena beberapa di antaranya dapat memicu efek paradoks yang terdiri dari penguatan dan peningkatan frekuensi kejang epilepsi. Hal ini terjadi jika obat tersebut digunakan sebagai bagian dari monoterapi.
Untuk mengatasi ngiler, biasanya digunakan dua metode: pengobatan (obat yang menekan produksi air liur) dan pembedahan, yang melibatkan penanaman kembali saluran air liur. Namun dalam kasus SA, metode ini dianggap tidak efektif, dan masalahnya masih terbuka. Orang tua dan mereka yang merawat pasien tersebut harus memberi perhatian khusus pada masalah ini, karena pasien sendiri biasanya tidak dapat mengendalikan ngiler, dan beberapa tidak dapat mengurus diri sendiri.
Masalah lainnya adalah durasi tidur yang pendek. Sering kali anak-anak dengan sindrom Angelman tidur tidak lebih dari 5 jam, yang berdampak negatif pada fungsi seluruh tubuh. Anak-anak yang mudah bersemangat, aktif, yang menyukai permainan dan komunikasi (bahkan jika mereka mencoba membatasi diri pada metode non-verbal) akan terlihat lelah di siang hari. Agar dapat beristirahat dengan baik, tubuh membutuhkan tidur yang nyenyak dan penuh, tetapi justru inilah kendalanya.
Tampaknya obat penenang (fenotiazin dan antipsikotik atipikal) yang menenangkan sistem saraf seharusnya cukup untuk meningkatkan kualitas tidur pada pasien yang mudah terangsang. Namun dalam kasus AS, penggunaan obat-obatan tersebut sarat dengan munculnya efek negatif. Oleh karena itu, dokter tetap lebih memilih pil tidur yang ringan, seperti Melatonin (obat hormonal alami yang berbahan dasar hormon tidur), yang diberikan kepada pasien satu jam sebelum tidur dalam jumlah 1 tablet, dan Difenhidramin. Frekuensi pemberian dan dosisnya ditentukan oleh dokter tergantung pada kondisi dan usia pasien.
Terkadang pasien dengan sindrom Angelman mengalami masalah pencernaan dan tinja. Anda dapat memperbaiki tinja dengan obat pencahar (sebaiknya yang herbal).
Atau Anda dapat mendekati masalah ini secara berbeda, seperti yang dilakukan oleh dokter Amerika, berdasarkan beberapa metode pengobatan autisme, karena banyak gejala khas AS juga merupakan ciri autisme (impulsivitas, gerakan tak terkendali, tindakan berulang, kurangnya perhatian, masalah komunikasi, dll.). Telah dicatat bahwa pengenalan hormon sekretin, yang menormalkan pencernaan dan tinja, memiliki efek positif pada perhatian pasien, dan oksitosin membantu meningkatkan kemampuan kognitif dan memori anak, serta memperbaiki perilaku.
Memang, hormon saja tidak cukup, terutama jika menyangkut anak-anak. Pada sindrom Angelman, terapi perilaku, bekerja sama dengan psikolog dan terapis wicara (mengajarkan metode komunikasi non-verbal dan bahasa isyarat) diindikasikan. Pendidikan anak-anak tersebut harus didasarkan pada program individual dengan partisipasi guru yang terlatih khusus, psikolog, dan orang tua. Sayangnya, ini tidak mungkin dilakukan di mana-mana, dan keluarga dibiarkan sendiri dengan masalah mereka.
Karena banyak pasien muda dengan AS menderita tonus otot rendah dan masalah sendi, banyak perhatian diberikan pada fisioterapi. Paling sering, dokter menggunakan aplikasi parafin, elektroforesis, dan terapi magnetik.
Pijat tonik aktif dan latihan khusus untuk latihan fisik terapeutik akan membantu anak yang sakit untuk berdiri tegak dan berjalan dengan percaya diri setelah beberapa saat. Senam air sangat berguna dalam hal ini, yang direkomendasikan untuk SA di air dingin. Senam air meningkatkan tonus otot dan mengajarkan anak untuk mengendalikan tubuhnya dan mengoordinasikan gerakan.
Pengobatan antikonvulsan
Gejala sindrom Angelman yang paling berbahaya adalah kejang yang mirip dengan epilepsi. Gejala ini terjadi pada 80% pasien, yang berarti bahwa semuanya perlu diberi resep pengobatan antikonvulsan yang efektif.
Pengobatan kejang epilepsi dilakukan dengan bantuan vitamin dan antikonvulsan. Pada sindrom Angelman, disertai dengan sindrom kejang, vitamin golongan B, serta vitamin C, D, dan E akan bermanfaat. Namun, meresepkan terapi vitamin sendiri dalam kasus ini sangat berbahaya, karena asupan vitamin yang tidak terkontrol dapat mengurangi efektivitas obat antiepilepsi dan memicu kejang baru yang lebih parah dan berkepanjangan.
Pemilihan obat antikonvulsan dan pemberian dosis efektifnya juga harus dilakukan oleh dokter spesialis. Ia juga yang memutuskan apakah satu obat sudah cukup atau apakah pasien harus mengonsumsi 2 obat atau lebih dalam jangka waktu lama.
Bagi sebagian besar pasien, dokter meresepkan obat asam valproat (Asam Valproat, Depakine, Convulex, Valparin, dll.), yang mencegah kejang dan memperbaiki suasana hati dan kondisi mental pasien.
Asam valproat tersedia dalam bentuk tablet, sirup, dan larutan injeksi. Obat yang paling populer adalah obat lepas lambat "Depakine" dalam bentuk tablet dan larutan untuk pemberian intravena. Dosis obat ditentukan oleh dokter secara individual, tergantung pada berat badan, usia, dan kondisi pasien.
Obat ini diminum saat makan 2 hingga 3 kali sehari. Dosis harian rata-rata adalah 20-30 mg per 1 kilogram berat badan pasien, dosis maksimum adalah 50 mg/kg per hari.
Kontraindikasi penggunaan. Jangan gunakan jika terjadi disfungsi hati dan pankreas, diatesis hemoragik, hepatitis, porfiria, dan hipersensitivitas terhadap obat.
Efek sampingnya meliputi tremor tangan, gangguan pencernaan dan tinja, serta perubahan berat badan.
"Topiramate" juga merupakan obat pilihan untuk SA. Obat ini diproduksi dalam bentuk tablet dan digunakan baik sebagai bagian dari monoterapi maupun dalam kombinasi dengan obat lain.
Cara pemberian dan dosis. Tablet diminum secara oral tanpa memperhatikan asupan makanan. Dosis harian awal untuk orang dewasa adalah 25-50 mg, untuk anak-anak - 0,5-1 mg/kg. Setiap minggu, dosis ditingkatkan sesuai dengan petunjuk dokter.
Obat ini tidak boleh dikonsumsi selama kehamilan dan menyusui, serta dalam kasus hipersensitivitas terhadap komponen-komponennya. Obat ini memiliki banyak efek samping yang berbeda.
Obat-obatan yang mungkin diresepkan dokter untuk sindrom Angelman: Clomazepam, Rivotril, Lamotrigine, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra, dll.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Obat tradisional dan homeopati
Obat tradisional, seperti sediaan homeopati, tentu saja relatif aman, tetapi efektivitas pengobatan tersebut untuk sindrom Angelman dapat dianggap kontroversial.
Meskipun pengobatan tradisional masih dapat membantu dalam beberapa hal. Kita berbicara tentang menghentikan kejang epilepsi. Dalam hal ini, pengobatan herbal bisa sangat efektif.
Efek yang baik diberikan oleh ramuan obat yang berbahan dasar peony, licorice, dan duckweed (komponennya dikonsumsi dalam jumlah yang sama). Ramuan tersebut perlu digiling menjadi tepung. Setelah 2 minggu sejak mulai mengonsumsinya, Anda dapat melihat penurunan frekuensi kejang yang signifikan.
Rebusan lavender (1 sendok teh per gelas air mendidih) juga bermanfaat untuk mengatasi kram. Campuran direbus selama 5 menit dan didiamkan selama setengah jam. Obat diminum pada malam hari selama 14 hari.
Infus motherwort dalam air (atau alkohol) dianggap efektif untuk kejang epilepsi.
Dari sediaan homeopati untuk mencegah kejang pada sindrom Angelman, Anda dapat menggunakan obat-obatan yang berbahan dasar chamomile dan motherwort, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Namun, perlu diingat bahwa hanya dokter homeopati yang dapat meresepkan dosis obat yang efektif dan aman dalam setiap kasus tertentu.
Pencegahan
Seperti yang mungkin sudah dipahami pembaca, kedokteran belum mampu mencegah mutasi gen dan kelainan kromosom lainnya, serta memperbaiki situasi tersebut. Hal ini dapat terjadi pada siapa saja, karena anak-anak dengan sindrom Angelman dilahirkan dari orang tua yang sehat, dan genetika, yang saat ini merupakan salah satu cabang kedokteran yang paling sedikit dipelajari, belum dapat menjelaskan hal ini.
Satu-satunya hal yang dapat dilakukan adalah mengambil pendekatan yang bertanggung jawab terhadap perencanaan kehamilan, mendaftar dan menjalani pemeriksaan tepat waktu. Namun sekali lagi, tindakan seperti itu akan lebih bersifat edukatif daripada preventif, seperti pemeriksaan lainnya. Namun, orang tua muda akan mengetahui terlebih dahulu apa yang harus dipersiapkan, dan jika jawabannya positif, mereka akan memutuskan apakah mereka dapat mengambil tanggung jawab seperti membesarkan anak yang sakit.
Ramalan cuaca
Prognosis untuk sindrom Angelman bergantung pada sifat kelainan kromosom dan ketepatan waktu deteksinya. Yang paling terdampak adalah anak-anak yang kromosom 15-nya mengandung "celah" dalam gen (delesi). Kemungkinan pasien tersebut dapat berjalan dan berbicara sangat rendah. Kasus-kasus lain dapat diperbaiki dengan pendekatan yang cermat dan kasih sayang kepada anak Anda.
Sayangnya, pasien seperti itu tidak akan dapat menjadi anggota masyarakat yang utuh, meskipun mereka jauh dari kata bodoh, mereka mengerti pembicaraan dan artinya. Namun, mereka akan mengalami masalah komunikasi selama sisa hidup mereka. Pasien dapat diajarkan bahasa isyarat sejak kecil, tetapi mereka tidak dapat dipaksa untuk berkomunikasi menggunakan kata-kata. Kosakata pasien yang "berbicara" terbatas pada kata-kata minimum yang digunakan dalam kehidupan sehari-hari (5-15 kata).
Mengenai harapan hidup dan kesehatan umum pasien dengan sindrom Angelman, angka-angka di sini berfluktuasi di sekitar nilai rata-rata. Di masa dewasa, pasien sebagian besar menghadapi masalah kesehatan seperti skoliosis dan obesitas, yang, dengan pendekatan pengobatan yang tepat, tidak mengancam jiwa.