Ahli medis artikel

Publikasi baru
Sindrom Usher
Terakhir ditinjau: 04.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
Sindrom Usher adalah penyakit keturunan yang bermanifestasi sebagai ketulian total sejak lahir, serta kebutaan progresif seiring bertambahnya usia. Kehilangan penglihatan dikaitkan dengan retinitis pigmentosa, suatu proses degenerasi pigmentasi retina. Banyak orang dengan sindrom Usher juga memiliki masalah keseimbangan yang parah.
Epidemiologi
Berkat penelitian tersebut, dapat dipastikan bahwa sindrom Usher memengaruhi sekitar 8% dari anak-anak tuli-bisu yang diperiksa (tes dilakukan di lembaga khusus untuk orang tuli-bisu). Retinitis pigmentosa ditemukan pada 6-10% pasien yang menderita tuli bawaan, yang pada gilirannya ditemukan pada sekitar 30% orang dengan penyakit retina pigmentosa.
Diperkirakan penyakit ini menyerang sekitar 3-10 orang dari 100 ribu orang di seluruh dunia. Penyakit ini dapat menyerang wanita dan pria secara seimbang. Sekitar 5-6% populasi dunia menderita sindrom ini. Sekitar 10% dari semua kasus tuli berat pada anak-anak terjadi karena sindrom Usher tipe I dan II.
Di Amerika Serikat, tipe 1 dan 2 merupakan tipe yang paling umum. Secara keseluruhan, kedua tipe ini mencakup sekitar 90 hingga 95 persen dari semua kasus sindrom Usher pada anak-anak.
Penyebab Sindrom Usher
Sindrom Usher tipe I, II, dan III memiliki penyebab resesif autosomal, sedangkan tipe IV dianggap sebagai kelainan kromosom X. Penyebab kebutaan dan ketulian yang terjadi pada sindrom ini belum diteliti secara memadai. Diduga penderita penyakit ini hipersensitif terhadap komponen yang dapat merusak struktur DNA. Selain itu, penyakit ini mungkin terkait dengan gangguan sistem imun, tetapi pada kasus ini belum ada gambaran pasti mengenai proses ini.
Pada tahun 1989, kelainan kromosom pertama kali diidentifikasi pada pasien dengan penyakit tipe II, yang mungkin di masa mendatang mengarah pada cara untuk mengisolasi gen yang menyebabkan sindrom tersebut. Mungkin juga memungkinkan untuk mengidentifikasi gen-gen ini pada pembawa dan mengembangkan tes genetik prenatal khusus.
 [ 8 ]
[ 8 ]
Faktor risiko
Sindrom ini diwariskan jika kedua orang tua terkena, yaitu diwariskan melalui tipe resesif. Seorang anak juga dapat mewarisi penyakit ini jika kedua orang tuanya merupakan pembawa gen tersebut. Jika kedua orang tua di masa depan memiliki gen ini, maka kemungkinan memiliki bayi dengan sindrom ini adalah 1 banding 4. Seseorang yang hanya memiliki satu gen untuk sindrom ini dianggap sebagai pembawa, tetapi tidak memiliki gejala gangguan tersebut. Saat ini, belum mungkin untuk menentukan apakah seseorang memiliki gen untuk penyakit ini.
Bila seorang anak dilahirkan dari orangtua yang salah satu tidak memiliki gen tersebut, maka kemungkinan ia akan mewarisi sindrom tersebut sangat rendah, tetapi ia pasti akan menjadi pembawa.
Gejala Sindrom Usher
Gejala sindrom Usher meliputi kehilangan pendengaran dan penumpukan sel berpigmen yang tidak normal pada struktur mata. Pasien kemudian mengalami degenerasi retina, yang menyebabkan penurunan penglihatan dan akhirnya kehilangan penglihatan pada kasus yang paling parah.
Gangguan pendengaran sensorineural bisa ringan atau total dan biasanya tidak memburuk sejak lahir. Namun, penyakit pigmen retina dapat mulai berkembang di masa kanak-kanak atau setelahnya. Hasil pengujian menunjukkan bahwa ketajaman penglihatan sentral dapat dipertahankan selama bertahun-tahun, bahkan ketika penglihatan tepi memburuk (kondisi yang disebut "penglihatan terowongan").
Ini adalah manifestasi utama penyakit, yang kadang-kadang dapat disertai oleh gangguan lain, seperti psikosis dan gangguan mental lainnya, masalah pada telinga bagian dalam dan/atau katarak.
Formulir
Selama penelitian, 3 jenis penyakit ini diidentifikasi, serta bentuk ke-4, yang cukup langka.
Tipe I penyakit ini ditandai dengan ketulian total bawaan, serta gangguan keseimbangan. Sering kali, anak-anak seperti itu mulai berjalan hanya pada usia 1,5 tahun. Penurunan penglihatan biasanya dimulai pada usia 10 tahun, dan perkembangan terakhir dari keadaan rabun senja dimulai pada usia 20 tahun. Anak-anak dengan jenis penyakit ini dapat mengalami penurunan penglihatan tepi secara progresif.
Pada penyakit tipe II, terjadi ketulian sedang atau bawaan. Dalam kasus ini, penurunan ketulian parsial sering kali tidak terjadi lagi. Retinitis pigmentosa mulai berkembang sekitar akhir masa remaja atau setelah 20 tahun. Perkembangan rabun senja biasanya dimulai pada usia 29-31 tahun. Gangguan ketajaman penglihatan pada kasus patologi tipe II umumnya berkembang sedikit lebih lambat daripada pada tipe I.
Tipe III penyakit ini ditandai dengan hilangnya pendengaran secara progresif, biasanya dimulai selama masa pubertas, serta perkembangan bertahap selama periode yang sama (sedikit lebih lambat dari hilangnya pendengaran) retinitis pigmentosa, yang dapat menjadi faktor dalam perkembangan kebutaan progresif.
Manifestasi patologi tipe IV terutama terjadi pada pria. Dalam kasus ini, gangguan progresif dan kehilangan pendengaran dan penglihatan juga diamati. Bentuk ini sangat langka dan biasanya memiliki sifat kromosom X.
Diagnostik Sindrom Usher
Diagnosis sindrom Usher dibuat berdasarkan kombinasi ketulian mendadak dan kehilangan penglihatan progresif yang diamati pasien.
Tes
Tes genetik khusus mungkin dipesan untuk mendeteksi mutasi.
Sebelas lokus genetik telah ditemukan yang dapat menyebabkan perkembangan sindrom Usher, dan sembilan gen telah diidentifikasi yang pasti menjadi penyebab gangguan tersebut:
- Tipe 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Tipe 2: ush2a, VLGR1, WHRN.
- Sindrom Usher tipe 3: USH3A.
Para ilmuwan di NIDCD, bersama dengan rekan-rekan dari berbagai universitas di New York dan Israel, telah mengidentifikasi mutasi yang disebut R245X pada gen Pcdh15 yang menjadi penyebab sebagian besar sindrom Usher tipe 1 pada populasi Yahudi.
Untuk mengetahui lebih lanjut tentang laboratorium yang melaksanakan uji klinis, kunjungi https://www.genetests.org dan cari direktori laboratorium dengan kata kunci "sindrom Usher".
Untuk mempelajari tentang uji klinis yang ada yang mencakup pengujian genetik untuk sindrom Usher, kunjungi https://www.clinicaltrials.gov dan cari "sindrom Usher" atau "pengujian genetik sindrom Usher."
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnostik instrumental
Ada beberapa metode diagnostik instrumental:
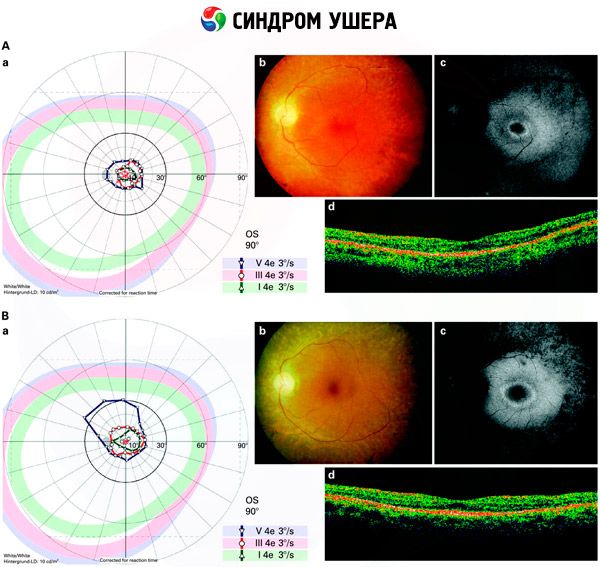
- Pemeriksaan fundus untuk mendeteksi adanya bintik pigmen pada retina, serta penyempitan pembuluh retina;
- Elektroretinogram, yang memungkinkan untuk mendeteksi deviasi degeneratif awal pada retina mata. Ini menunjukkan hilangnya jalur elektroradiografi;
- Elektronistagmogram (ENG) mengukur gerakan mata tak sadar yang dapat mengindikasikan adanya ketidakseimbangan.
- Audiometri, yang digunakan untuk menentukan adanya ketulian dan tingkat keparahannya.
Perbedaan diagnosa
Sindrom Usher harus dibedakan dari beberapa gangguan serupa.
Sindrom Hallgren, yang ditandai dengan kehilangan pendengaran bawaan dan kehilangan penglihatan progresif (katarak dan nistagmus juga berkembang). Gejala tambahan meliputi ataksia, gangguan psikomotorik, psikosis, dan retardasi mental.
Sindrom Alstrom, yaitu penyakit keturunan yang menyebabkan degenerasi retina, yang mengakibatkan hilangnya penglihatan sentral. Sindrom ini dikaitkan dengan obesitas pada anak-anak. Pada saat yang sama, diabetes melitus dan gangguan pendengaran mulai berkembang setelah 10 tahun.
Rubella pada ibu hamil di trimester pertama dapat menyebabkan berbagai kelainan pada perkembangan anak. Di antara konsekuensi kelainan tersebut adalah gangguan pendengaran, serta (atau) masalah penglihatan, dan di samping itu, berbagai cacat perkembangan.
Siapa yang harus dihubungi?
Pengobatan Sindrom Usher
Saat ini belum ada obat untuk sindrom Usher. Oleh karena itu, terapi dalam kasus ini terutama terdiri dari memperlambat proses kehilangan penglihatan, serta mengompensasi kehilangan pendengaran. Metode pengobatan yang mungkin meliputi:
- Mengonsumsi vitamin A (sebagian dokter mata yakin bahwa dosis tinggi vitamin A palmitat dapat memperlambat, tetapi tidak menghentikan, perkembangan retinitis pigmentosa);
- Implantasi perangkat elektronik khusus ke telinga pasien (alat bantu dengar, implan koklea.
Dokter mata menyarankan agar kebanyakan orang dewasa dengan bentuk umum retinitis pigmentosa mengonsumsi 15.000 IU (unit internasional) vitamin A palmitat setiap hari di bawah pengawasan. Karena orang dengan sindrom Usher tipe 1 tidak disertakan dalam penelitian, dosis tinggi vitamin A tidak disarankan untuk kelompok pasien ini. Orang yang mempertimbangkan untuk mengonsumsi vitamin A harus mendiskusikan pilihan pengobatan ini dengan dokter mereka. Rekomendasi lain untuk pilihan pengobatan ini meliputi:
- Mengubah pola makan Anda untuk memasukkan makanan tinggi vitamin A.
- Wanita yang berencana untuk hamil sebaiknya berhenti mengonsumsi vitamin A dosis tinggi tiga bulan sebelum mereka berencana untuk hamil karena meningkatnya risiko cacat lahir.
- Wanita yang sedang hamil sebaiknya berhenti mengonsumsi vitamin A dosis tinggi karena dapat meningkatkan risiko cacat lahir.
Penting juga untuk menyesuaikan anak tersebut dengan kehidupan sosial. Ini memerlukan bantuan guru pendidikan khusus dan psikolog. Jika pasien mulai mengalami kehilangan penglihatan secara progresif, ia harus diajari menggunakan bahasa isyarat.
Ramalan cuaca
Sindrom Usher memiliki prognosis yang tidak baik. Bidang penglihatan dan ketajamannya mulai memburuk dalam kurun waktu 20-30 tahun pada sebagian besar pasien dengan penyakit ini, apa pun jenisnya. Dalam beberapa kasus, terjadi kehilangan penglihatan bilateral secara total. Kehilangan pendengaran, yang selalu disertai dengan kebisuan, dengan sangat cepat berkembang menjadi kehilangan pendengaran bilateral secara total.