Ahli medis artikel

Publikasi baru
Sindrom Treacher Collins
Terakhir ditinjau: 04.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.

Gangguan intrauterin pada proses perkembangan tulang menyebabkan kelainan kraniofasial yang serius, dan salah satu jenis patologi tersebut adalah sindrom Treacher Collins (TCS) atau mandibulofasial, yaitu disostosis maksilofasial.
Kode penyakit menurut ICD 10: kelas XVII (anomali kongenital, deformasi dan kelainan kromosom), Q75.4 - disostosis mandibulofasial.
Penyebab Sindrom Treacher Collins
Sindrom ini dinamai menurut dokter mata terkemuka Inggris Edward Treacher Collins, yang menggambarkan ciri-ciri utama patologi lebih dari seratus tahun yang lalu. Namun, dokter Eropa lebih sering menyebut jenis kelainan tulang wajah dan rahang ini sebagai penyakit atau sindrom Franceschetti - berdasarkan penelitian ekstensif dokter mata Swiss Adolf Franceschetti, yang memperkenalkan istilah "disostosis mandibulofasial" pada pertengahan abad lalu. Di kalangan medis, nama sindrom Franceschetti-Collins juga digunakan.
Sindrom Treacher Collins disebabkan oleh mutasi pada gen TCOF1 (pada lokus kromosom 5q31.3-33.3), yang mengkode fosfoprotein nukleolus yang bertanggung jawab atas pembentukan bagian kraniofasial embrio manusia. Akibat penurunan jumlah protein ini secara prematur, biogenesis dan fungsi rRNA terganggu. Menurut ahli genetika dari program penelitian Genom Manusia, proses ini menyebabkan pengurangan proliferasi sel embrio pada puncak saraf - tonjolan di sepanjang alur saraf, yang menutup menjadi tabung saraf selama perkembangan embrio.
Pembentukan jaringan wajah terjadi karena transformasi dan diferensiasi sel-sel bagian atas (kepala) dari krista saraf, yang bermigrasi sepanjang tabung saraf ke area lengkung brankial pertama dan kedua embrio. Dan kekurangan sel-sel ini menyebabkan deformasi kraniofasial. Periode kritis untuk terjadinya anomali adalah dari 18 hingga 28 hari setelah pembuahan. Setelah selesainya migrasi sel-sel krista saraf (pada minggu keempat kehamilan), hampir semua jaringan mesenkim longgar di area wajah terbentuk, yang kemudian (dari 5 hingga 8 minggu) berdiferensiasi menjadi jaringan rangka dan ikat dari semua bagian wajah, leher, laring, telinga (termasuk telinga bagian dalam) dan gigi masa depan.
Patogenesis
Patogenesis sindrom Treacher Collins sering kali bersifat familial, dan anomali tersebut diwariskan secara dominan autosomal, meskipun ada beberapa kasus transmisi defek resesif autosomal (dengan mutasi pada gen lain, khususnya POLR1C dan POLR1D). Hal yang paling tidak dapat diprediksi tentang disostosis maksilofasial adalah bahwa mutasi tersebut diwariskan kepada anak-anak hanya pada 40-48% kasus. Artinya, pada 52-60% pasien, penyebab sindrom Treacher Collins tidak terkait dengan adanya anomali dalam keluarga, dan diyakini bahwa patologi tersebut terjadi sebagai akibat dari mutasi gen sporadis de novo. Kemungkinan besar, mutasi baru merupakan konsekuensi dari efek teratogenik pada janin selama kehamilan.
Di antara penyebab teratogenik sindrom ini, para ahli menyebutkan dosis besar etanol (etil alkohol), radiasi, asap rokok, sitomegalovirus dan toksoplasma, serta herbisida berbasis glifosat (Roundal, Glyfor, Tornado, dll.). Dan daftar faktor iatrogenik mencakup obat jerawat dan seborrhea dengan asam 13-cis-retinoat (Isotretinoin, Accutane); obat antikonvulsan Phenytoin (Dilantin, Epanutin); obat psikotropika Diazepam, Valium, Relanium, Seduxen.
Gejala Sindrom Treacher Collins
Untuk sebagian besar, tanda-tanda klinis disostosis mandibulofasial dan tingkat ekspresinya bergantung pada karakteristik manifestasi mutasi gen. Dan tanda-tanda pertama anomali ini dalam banyak kasus terlihat pada anak segera setelah lahir: wajah dengan sindrom Treacher Collins memiliki penampilan yang khas. Selain itu, anomali morfologis biasanya bilateral dan simetris.
Gejala sindrom Treacher Collins yang paling jelas adalah:
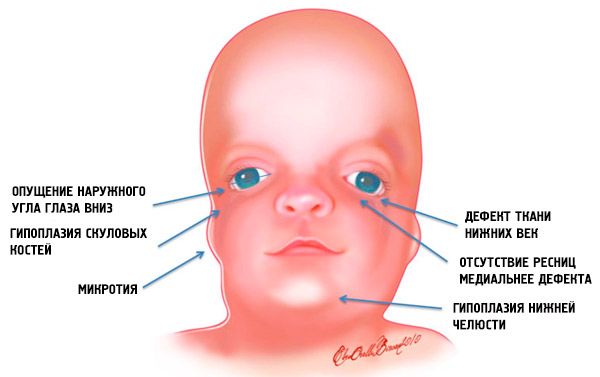
- keterbelakangan (hipoplasia) tulang wajah tengkorak: zygomatik, proses zygomatik tulang frontal, lempeng pterigoid lateral, sinus paranasal, rahang bawah dan tonjolan epifisis tulang (kondilus);
- keterbelakangan tulang rahang bawah (mikrognathia) dan sudut mandibula yang lebih tumpul dari biasanya;
- hidung berukuran normal, tetapi tampak besar karena hipoplasia lengkungan supersiliaris dan kurang berkembangnya atau tidak adanya lengkungan zygomatik di daerah temporal;
- celah mata mengarah ke bawah, yaitu bentuk mata tidak normal, sudut luar mata menurun ke bawah;
- cacat pada kelopak mata bawah (koloboma) dan sebagian tidak adanya bulu mata pada kelopak mata tersebut;
- daun telinga yang bentuknya tidak teratur dengan berbagai macam deviasi, antara lain letaknya di sudut rahang bawah, tidak adanya lobus, fistula buta antara tragus telinga dan sudut mulut, dan lain-lain;
- penyempitan atau penutupan (atresia) saluran pendengaran eksternal dan anomali tulang-tulang telinga tengah;
- tidak adanya atau hipoplasia kelenjar ludah parotis;
- hipoplasia faring (penyempitan faring dan saluran udara);
- tidak menyatunya langit-langit keras (langit-langit sumbing), dan juga tidak adanya, pemendekan atau imobilitas langit-langit lunak.
Kelainan anatomi seperti itu dalam semua kasus memiliki komplikasi. Yaitu gangguan pendengaran fungsional berupa kehilangan pendengaran konduktif atau tuli total; gangguan penglihatan akibat pembentukan bola mata yang tidak tepat; cacat pada langit-langit mulut yang menyebabkan kesulitan makan dan menelan. Ada kelainan oklusi gigi (maloklusi) yang terkait dengan cacat rahang, yang pada gilirannya menyebabkan masalah mengunyah dan artikulasi. Patologi langit-langit lunak menjelaskan suara sengau.
Komplikasi dan konsekuensinya
Konsekuensi dari anomali maksilofasial pada sindrom Treacher Collins adalah bahwa saat lahir kemampuan intelektual anak normal, tetapi karena cacat pendengaran dan gangguan lainnya, keterbelakangan mental sekunder diamati.
Selain itu, anak-anak dengan cacat seperti itu sangat merasakan rendah diri dan menderita, yang berdampak negatif pada sistem saraf dan jiwa mereka.
Diagnostik Sindrom Treacher Collins
Diagnosis sindrom Treacher Collins pascanatal pada dasarnya didasarkan pada tanda-tanda klinis. Disostosis kraniofasial mudah diidentifikasi ketika sindrom tersebut sepenuhnya ekspresif, tetapi ketika gejala patologi yang diekspresikan secara minimal muncul, masalah dalam menegakkan diagnosis yang tepat dapat muncul.
Dalam kasus ini, perhatian khusus harus diberikan pada penilaian semua fungsi yang terkait dengan anomali, terutama yang memengaruhi pernapasan (karena risiko sleep apnea). Efektivitas pemberian makan dan saturasi oksigen hemoglobin juga harus dinilai dan dipantau.
Nantinya, pada hari ke 5-6 pasca kelahiran, tingkat kerusakan pendengaran perlu diketahui dengan menggunakan pemeriksaan audiologi yang dapat dilakukan di rumah sakit bersalin.
Pemeriksaan ditentukan, di mana diagnostik instrumental dilakukan dengan fluoroskopi dismorfologi kraniofasial; pantomografi (rontgen panoramik pada struktur tulang tengkorak wajah); tomografi terkomputasi kranial penuh dalam berbagai proyeksi; CT atau MRI otak untuk menentukan keadaan liang pendengaran internal.
Diagnosis paling awal – prenatal – untuk kelainan maksilofasial dengan adanya sindrom Treacher Collins dalam riwayat keluarga dapat dilakukan melalui biopsi vili korionik pada usia kehamilan 10-11 minggu (prosedur ini berisiko keguguran dan infeksi rahim).
Tes darah juga diambil dari anggota keluarga; pada usia kehamilan 16-17 minggu, cairan ketuban dianalisis (amniosentesis transabdominal); pada usia kehamilan 18-20 minggu, fetoskopi dilakukan dan darah diambil dari pembuluh janin plasenta.
Namun yang paling sering, USG digunakan dalam diagnosis prenatal sindrom ini pada janin (pada usia kehamilan 20-24 minggu).
Tes apa yang dibutuhkan?
Perbedaan diagnosa
Metode yang sama digunakan oleh para spesialis ketika diagnostik diferensial dibutuhkan untuk mengenali sindrom Treacher Collins yang ringan dan membedakannya dari anomali kongenital tulang kraniofasial lainnya, khususnya: sindrom Apert, Crouzon, Nager, Peters-Hewels, Hellermann-Steph, serta mikrosomia hemifasial (sindrom Goldenhar), hipertelorisme, fusi prematur sutura kranial (kraniosinostosis) atau gangguan fusi tulang wajah (kraniosinostosis).
Siapa yang harus dihubungi?
Pengobatan Sindrom Treacher Collins
Seperti dalam semua kasus cacat bawaan yang ditentukan secara genetik, pengobatan sindrom Treacher Collins yang parah hanya bersifat paliatif, karena tidak ada metode terapi untuk patologi semacam itu. Spektrum dan tingkat deformasi pada sindrom ini sangat luas dan, oleh karena itu, sifat dan intensitas intervensi medis juga memiliki banyak pilihan.
Alat bantu dengar digunakan untuk mengoreksi dan meningkatkan pendengaran, dan sesi terapi wicara digunakan untuk meningkatkan kemampuan bicara.
Intervensi bedah diperlukan pada usia dini pada kasus penyempitan saluran napas yang parah (dilakukan trakeostomi) dan laring (dilakukan gastrostomi untuk makan). Koreksi bedah pada langit-langit juga mungkin diperlukan.
Operasi pemanjangan mandibula dilakukan pada usia 2-3 tahun atau lebih. Rekonstruksi jaringan lunak meliputi koreksi koloboma kelopak mata bawah dan operasi plastik aurikular.
Pencegahan
Ramalan cuaca
Bagaimana prognosis untuk patologi ini? Tergantung pada tingkat deformasi dan intensitas gejala. Sindrom Treacher Collins adalah diagnosis seumur hidup.
 [ 25 ]
[ 25 ]