Ahli medis artikel

Publikasi baru
Penyakit Gigi Charcot-Marie.
Terakhir ditinjau: 12.07.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
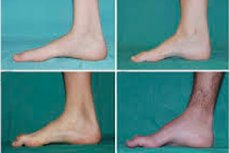
Atrofi otot peroneal, sindrom atau penyakit Charcot-Marie-Tooth adalah sekelompok penyakit keturunan kronis dengan kerusakan pada saraf perifer.
Menurut ICD-10, pada bagian penyakit sistem saraf, kode untuk penyakit ini adalah G60.0 (neuropati motorik dan sensorik herediter). Penyakit ini juga termasuk dalam daftar penyakit langka.
Epidemiologi
Menurut statistik klinis, prevalensi semua jenis penyakit Charcot-Marie-Tooth per 100 ribu populasi adalah 19 kasus (menurut sumber lain, satu kasus per 2,5-10 ribu populasi).
CMT tipe 1 mencakup sekitar dua pertiga kasus (satu kasus per 5.000 hingga 7.000 populasi), dan hampir 70% di antaranya terkait dengan duplikasi gen PMP22. Lebih dari 1,2 juta orang di seluruh dunia menderita jenis penyakit ini.
Insiden CMT tipe 4 diperkirakan 1-5 kasus per 10.000 anak. [ 1 ]
Penyebab Penyakit Gigi Charcot-Marie
Menurut klasifikasi sindrom polineuropati, atrofi otot peroneal (fibular), amiotrofi saraf Charcot-Marie-Tooth atau penyakit Charcot-Marie-Tooth (disingkat CMT) mengacu pada polineuropati motorik-sensorik yang ditentukan secara genetik. [ 2 ]
Artinya, penyebab terjadinya adalah mutasi genetik. Dan tergantung pada sifat penyimpangan genetik, jenis atau macam utama sindrom ini dibedakan: demielinasi dan akson. Kelompok pertama meliputi penyakit Charcot-Marie-Tooth tipe 1 (CMT1), yang terjadi karena duplikasi gen PMP22 pada kromosom 17, yang mengkode protein mielin perifer transmembran 22. Akibatnya, terjadi demielinasi segmental selubung akson (proses sel saraf) dan penurunan kecepatan konduksi sinyal saraf. Selain itu, mutasi mungkin terjadi pada beberapa gen lainnya.
Bentuk aksonnya adalah penyakit Charcot-Marie-Tooth tipe 2 (CMT2), yang memengaruhi akson itu sendiri dan dikaitkan dengan perubahan patologis pada gen MFN2 di lokus 1p36.22, yang mengkode protein membran mitofusin-2, yang diperlukan untuk fusi mitokondria dan pembentukan jaringan mitokondria fungsional di dalam sel saraf perifer. Ada lebih dari selusin subtipe CMT2 (dengan mutasi pada gen tertentu).
Perlu dicatat bahwa saat ini lebih dari seratus gen telah diidentifikasi yang kerusakannya, yang diwariskan, menyebabkan berbagai subtipe penyakit Charcot-Marie-Tooth. Misalnya, mutasi pada gen RAB7 menyebabkan CMT tipe 2B; perubahan gen SH3TC2 (yang mengkode salah satu protein membran sel Schwann) menyebabkan CMT tipe 4C, yang muncul pada masa kanak-kanak dan ditandai dengan demielinasi neuron motorik dan sensorik (ada selusin setengah bentuk tipe 4 penyakit ini).
CMT tipe 3 yang langka (disebut sindrom Dejerine-Sottas) mulai berkembang pada masa kanak-kanak dan disebabkan oleh mutasi pada gen PMP22, MPZ, EGR2, dan gen lainnya.
Bila CMT tipe 5 terjadi pada usia 5-12 tahun, yang terjadi bukan hanya neuropati motorik (berupa paraparesis spastik pada ekstremitas bawah), tetapi juga kerusakan saraf optik dan pendengaran.
Kelemahan otot dan atrofi optik (dengan hilangnya penglihatan) serta masalah keseimbangan merupakan gejala khas CMT tipe 6. Dan pada penyakit Charcot-Marie-Tooth tipe 7 tidak hanya terjadi neuropati motorik-sensorik, tetapi juga penyakit retina dalam bentuk retinitis pigmentosa.
Lebih umum di antara laki-laki, CMT terkait-X atau penyakit Charcot-Marie-Tooth dengan tetraparesis anggota badan (kelemahan gerakan kedua lengan dan kaki) adalah jenis demielinasi dan diduga disebabkan oleh mutasi pada gen GJB1 pada lengan panjang kromosom X, yang mengkode connexin 32, protein transmembran sel Schwann dan oligodendrosit yang mengatur transmisi sinyal saraf. [ 3 ]
Faktor risiko
Faktor risiko utama untuk CMT adalah riwayat penyakit dalam keluarga, yaitu pada kerabat dekat.
Menurut ahli genetika, jika kedua orang tua merupakan pembawa gen resesif autosomal untuk penyakit Charcot-Marie-Tooth, risiko memiliki anak yang akan mengidap penyakit ini adalah 25%. Dan risiko anak tersebut akan menjadi pembawa gen ini (tetapi tidak akan menunjukkan gejala apa pun) diperkirakan sebesar 50%.
Dalam kasus pewarisan terkait kromosom X (ketika gen yang bermutasi berada pada kromosom X wanita), ada risiko 50% bahwa ibu akan mewariskan gen tersebut kepada putranya, yang akan mengembangkan CMT. Penyakit ini mungkin tidak terjadi ketika anak perempuan lahir, tetapi anak laki-laki (cucu) dari anak perempuan tersebut dapat mewarisi gen yang rusak, dan penyakit tersebut akan berkembang.
Patogenesis
Pada semua jenis penyakit Charcot-Marie-Tooth, patogenesisnya disebabkan oleh kelainan herediter pada saraf tepi: motorik (gerakan) dan sensorik (sensitif).
Bila tipe CMT adalah demielinasi, maka kerusakan atau cacat pada selubung mielin yang melindungi akson saraf tepi menyebabkan perlambatan penghantaran impuls saraf pada susunan saraf tepi, antara otak, otot, dan organ sensorik.
Pada penyakit tipe akson, akson itu sendiri terpengaruh, yang berdampak negatif pada kekuatan sinyal saraf, sehingga tidak cukup untuk merangsang otot dan organ sensorik secara penuh.
Baca juga:
Bagaimana sindrom Charcot-Marie-Tooth ditularkan? Gen yang rusak dapat diwariskan secara autosomal dominan atau autosomal resesif.
Jenis yang paling umum, pewarisan dominan autosomal, terjadi ketika ada satu salinan gen yang bermutasi (dibawa oleh salah satu orang tua). Dan kemungkinan mewariskan CMT kepada setiap anak yang lahir diperkirakan sebesar 50%. [ 4 ]
Dengan pewarisan resesif autosom, dua salinan gen yang rusak (satu dari setiap orang tua yang tidak menunjukkan tanda-tanda penyakit apa pun) diperlukan agar penyakit dapat berkembang.
Pada 40-50% kasus, terjadi demielinasi herediter dominan autosomal, yaitu CMT tipe 1; pada 12-26% kasus, CMT akson, yaitu tipe 2. Dan pada 10-15% kasus, terjadi pewarisan terkait kromosom X. [ 5 ]
Gejala Penyakit Gigi Charcot-Marie
Biasanya, tanda-tanda pertama penyakit ini mulai muncul pada masa kanak-kanak dan remaja dan berkembang secara bertahap sepanjang hidup, meskipun sindrom ini mungkin baru diketahui kemudian. Kombinasi gejalanya bervariasi, dan laju perkembangan penyakit, serta tingkat keparahannya, tidak mungkin diprediksi.
Gejala khas tahap awal meliputi peningkatan kelelahan umum; penurunan tonus (kelemahan) otot-otot kaki, pergelangan kaki, dan tulang kering; kurangnya refleks. Hal ini membuat gerakan kaki menjadi sulit dan menyebabkan disbasia (gangguan gaya berjalan) dalam bentuk kaki yang terangkat lebih tinggi, sering kali disertai sering tersandung dan jatuh. Tanda-tanda penyakit Charcot-Marie-Tooth pada anak kecil dapat meliputi kecanggungan yang nyata dan kesulitan berjalan yang tidak sesuai dengan usia yang terkait dengan penurunan kaki bilateral. Kelainan bentuk kaki juga merupakan ciri khas: lengkungan tinggi (kaki berongga) atau kaki datar yang parah, jari-jari kaki melengkung (berbentuk palu).
Dalam kasus berjalan jinjit dengan latar belakang hipotonia otot, seorang ahli saraf mungkin menduga bahwa anak tersebut menderita CMT tipe 4, di mana anak mungkin tidak dapat berjalan pada masa remaja.
Seiring perkembangan penyakit, atrofi dan kelemahan otot menyebar ke ekstremitas atas, sehingga menyulitkan untuk melakukan keterampilan motorik halus dan melakukan tugas tangan normal. Sensasi sentuhan yang menurun dan kemampuan untuk merasakan panas dan dingin, serta mati rasa di kaki dan tangan, menunjukkan kerusakan pada akson saraf sensorik.
Pada penyakit Charcot-Marie-Tooth tipe 3 dan 6 yang terjadi pada masa kanak-kanak, diamati adanya ataksia sensorik (gangguan koordinasi gerakan dan keseimbangan), kedutan otot dan tremor, kerusakan saraf wajah, atrofi saraf optik dengan nistagmus, dan gangguan pendengaran.
Pada stadium lanjut, mungkin terjadi gemetar yang tidak terkendali (tremor) dan kram otot yang sering terjadi; masalah pergerakan dapat memicu timbulnya nyeri: otot, sendi, neuropatik.
Komplikasi dan konsekuensinya
Penyakit Charcot-Marie-Tooth dapat menimbulkan komplikasi dan konsekuensi seperti:
- terkilir dan patah tulang lebih sering terjadi;
- kontraktur yang terkait dengan pemendekan otot dan tendon periartikular;
- skoliosis (kelengkungan tulang belakang);
- masalah pernapasan – ketika serabut saraf yang mempersarafi otot diafragma rusak:
- hilangnya kemampuan untuk bergerak secara mandiri.
Diagnostik Penyakit Gigi Charcot-Marie
Diagnosis meliputi pemeriksaan klinis, anamnesis (termasuk riwayat keluarga), pemeriksaan neurologis dan sistemik.
Tes dilakukan untuk memeriksa rentang gerak, sensitivitas, dan refleks tendon. Konduksi saraf dapat dinilai menggunakan diagnostik instrumental – elektromiografi atau elektroneuromiografi. Ultrasonografi atau MRI mungkin juga diperlukan. [ 6 ]
Pengujian genetik atau DNA untuk mendeteksi mutasi genetik paling umum yang menyebabkan CMT dalam sampel darah terbatas karena pengujian DNA saat ini tidak tersedia untuk semua jenis penyakit. Untuk informasi lebih lanjut, lihat Pengujian genetik
Dalam beberapa kasus, biopsi saraf tepi (biasanya saraf sural) dilakukan.
Perbedaan diagnosa
Diagnosis banding mencakup neuropati perifer lainnya, distrofi otot Duchenne, sindrom mielopati dan miastenia, neuropati diabetik, mielopati pada sklerosis lateral multipel dan amiotrofik, sindrom Guillain-Barré, trauma pada saraf peroneal dan atrofinya (termasuk saat terjepit di antara diskus lumbal tulang belakang), kerusakan pada otak kecil atau talamus, serta efek samping kemoterapi (selama pengobatan dengan sitostatika seperti Vincristine atau Paclitaxel). [ 7 ]
Siapa yang harus dihubungi?
Pengobatan Penyakit Gigi Charcot-Marie
Saat ini, pengobatan untuk penyakit keturunan ini meliputi terapi latihan (yang bertujuan untuk memperkuat dan meregangkan otot); terapi okupasi (yang membantu pasien dengan kelemahan otot di tangan); dan penggunaan alat ortopedi untuk memudahkan berjalan. Jika perlu, obat pereda nyeri atau antikonvulsan diresepkan. [ 8 ]
Pada kasus kaki datar yang parah, osteotom dapat dilakukan, dan pada kasus deformasi tumit, koreksi bedah diindikasikan – artrodesis. [ 9 ]
Penelitian sedang dilakukan terhadap komponen genetik penyakit dan metode pengobatannya. Penggunaan sel induk, hormon tertentu, lesitin atau asam askorbat belum membuahkan hasil positif.
Namun berkat penelitian terkini, sesuatu yang baru mungkin memang muncul dalam pengobatan penyakit Charcot-Marie-Tooth dalam waktu dekat. Jadi, sejak 2014, perusahaan Prancis Pharnext telah mengembangkan, dan sejak pertengahan 2019, uji klinis telah dilakukan untuk obat PXT3003 untuk pengobatan CMT tipe 1 pada orang dewasa, yang menekan peningkatan ekspresi gen PMP22, meningkatkan mielinisasi saraf tepi, dan meringankan gejala neuromuskular.
Spesialis dari perusahaan medis Sarepta Therapeutics (AS) tengah berupaya menciptakan terapi gen untuk penyakit Charcot-Marie-Tooth tipe 1. Terapi ini akan menggunakan virus adeno-associated (AAV) yang tidak berbahaya dari genus Dependovirus dengan genom DNA untai tunggal linier, yang akan mentransfer gen NTF3 ke dalam tubuh, yang mengkode protein neurotrophin-3 (NT-3) yang diperlukan untuk berfungsinya sel saraf Schwann.
Helixmith akan memulai uji klinis terapi gen Engensis (VM202) yang dikembangkan Korea Selatan pada akhir tahun 2020 untuk mengobati gejala otot pada CMT tipe 1. [ 10 ]
Pencegahan
Pencegahan CMT dapat dilakukan dengan konseling genetika bagi calon orang tua, terutama jika salah satu pasangan memiliki riwayat keluarga dengan penyakit tersebut. Namun, kasus mutasi gen titik de novo telah teridentifikasi, yaitu tidak adanya penyakit tersebut dalam riwayat keluarga.
Selama kehamilan, kemungkinan penyakit Charcot-Marie-Tooth pada bayi di masa depan dapat diperiksa melalui biopsi vili korionik (dari usia kehamilan 10 hingga 13 minggu), serta analisis cairan ketuban (pada usia kehamilan 15-18 minggu).
Ramalan cuaca
Secara umum, prognosis untuk berbagai jenis penyakit Charcot-Marie-Tooth bergantung pada tingkat keparahan klinis, tetapi dalam semua kasus penyakit ini berkembang secara perlahan. Banyak pasien yang mengalami kecacatan, meskipun hal ini tidak mengurangi harapan hidup.