Ahli medis artikel

Publikasi baru
hamartoma
Terakhir ditinjau: 29.06.2025

Semua konten iLive ditinjau secara medis atau diperiksa fakta untuk memastikan akurasi faktual sebanyak mungkin.
Kami memiliki panduan sumber yang ketat dan hanya menautkan ke situs media terkemuka, lembaga penelitian akademik, dan, jika mungkin, studi yang ditinjau secara medis oleh rekan sejawat. Perhatikan bahwa angka dalam tanda kurung ([1], [2], dll.) Adalah tautan yang dapat diklik untuk studi ini.
Jika Anda merasa salah satu konten kami tidak akurat, ketinggalan zaman, atau dipertanyakan, pilih dan tekan Ctrl + Enter.
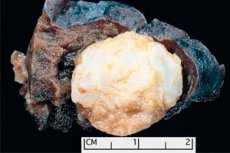
Pembentukan seperti tumor yang terlokalisasi di daerah anatomi mana pun yang diakibatkan oleh pertumbuhan jaringan jinak yang abnormal, dalam dunia kedokteran didefinisikan sebagai hamartoma (dari bahasa Yunani hamartia - kesalahan, cacat). [ 1 ]
Epidemiologi
Secara statistik, hamartoma mencakup 1,2% dari neoplasma jinak. Prevalensi hamartoma paru diperkirakan sekitar 0,25% dari populasi umum dan mencakup hingga 8% dari semua neoplasma paru. Sebagian besar hamartoma paru didiagnosis secara tidak sengaja pada pasien berusia 40 hingga 70 tahun, tetapi sangat jarang terjadi pada praktik pediatrik.
Secara umum, sebagian besar hamartoma didiagnosis pada pria, meskipun pada ginjal lebih umum terjadi pada wanita dan teridentifikasi pada usia paruh baya.
Sekitar 5% tumor payudara jinak adalah hamartoma, dan paling sering menyerang wanita berusia di atas 35 tahun.
80-90% lesi hamartomatosa otak dan lebih dari 50% hamartoma jantung dikaitkan dengan sklerosis tuberositas.
Penyebab hamartomas
Gamartoma termasuk kelainan bawaan dan merupakan formasi jinak yang terbentuk dari jaringan mesenkim yang berasal dari lapisan germinal. Penyebab kemunculannya dikaitkan dengan pembelahan sel yang tidak terkendali dari jaringan yang secara sitologis normal (jaringan ikat, otot polos, lemak atau tulang rawan), karakteristik lokasi anatomi tertentu, dan pertumbuhan fokal yang berlebihan selama embriogenesis hampir semua organ atau struktur anatomi.
Terjadinya beberapa hamartoma pada pasien yang sama sering disebut sebagai hamartomatosis atau hamartoma pleiotropik.
Tumor ini dapat muncul secara sporadis atau disertai adanya penyakit bawaan autosomal dominan tertentu serta sindrom yang ditentukan secara genetik.
Dalam banyak kasus, hamartoma terbentuk ketika penyakit genetik langka yang bersifat multisistemik - tuberous sclerosis - muncul segera setelah lahir, atau pada penyakit familial Recklinghausen - neurofibromatosis tipe 1. [ 2 ]
Faktor risiko
Faktor risiko utama untuk pembentukan hamartoma meliputi adanya apa yang disebut sindrom genetik poliposis hamartomatosa dalam riwayat pasien, termasuk:
- Sindrom hamartoma multipel - sindrom Cowden, di mana terbentuk multipel hamartoma asal ekto-, ento- dan mesodermal, diamati adanya poliposis gastrointestinal dan manifestasi mukokutan;
- Sindrom Peutz-Jeghers-Turen (ditandai dengan perkembangan polip hamartomatosa jinak di saluran pencernaan);
- sindrom Proteus;
- Sindrom Weil - poliposis juvenil pada usus besar;
- Sindrom Bannayan-Riley-Ruvalcaba, yang seperti sindrom Cowden, menghasilkan banyak hamartoma (polip hamartomatosa) pada usus;
- Sindrom Carney-Stratakis dan kompleks Carney.
Selain itu, hamartoma terbentuk pada pasien dengan sindrom Watson herediter dan dalam kasus sindrom Pallister-Hall yang terjadi secara sporadis atau kongenital dengan hamartoma hipotalamus dan polidaktili.
Patogenesis
Mekanisme peningkatan proliferasi jaringan germinal dengan pembentukan malformasi seperti tumor di berbagai organ dijelaskan oleh aberasi kromosom dan mutasi gen yang dapat terjadi secara spontan atau diwariskan.
Pada sklerosis tuberosa, mutasi pada gen TSC1 atau TSC2 - penekan tumor yang mencegah dan menghambat proliferasi berlebihan - pertumbuhan dan pembelahan sel yang terlalu cepat atau tidak terkendali - telah diidentifikasi. Dan pada neurofibromatosis tipe 1 dan sindrom Watson - mutasi garis keturunan gen penekan tumor mitokondria NF1.
Pada sindrom tumor hamartoma, yang menggabungkan sindrom Cowden, Protea, Bannayan-Riley-Ruvalcaba, dan poliposis juvenil, patogenesisnya dikaitkan dengan mutasi gen PTEN, yang mengkode enzim yang terlibat dalam pengaturan proliferasi dan dianggap sebagai gen penekan tumor.
Mutasi pada gen STK11 yang mengkodekan struktur dan fungsi salah satu enzim serin transmembran, yang mengurangi kemampuannya untuk menahan pembelahan sel, menyebabkan sindrom Peutz-Jeghers-Turen, dengan perkembangan polip usus dan lesi kulit berpigmen. Mutasi pada gen GLI3, faktor transkripsi yang terlibat dalam pembentukan jaringan intrauterin, telah diidentifikasi pada sindrom Pallister-Hall.
Dengan demikian, pertumbuhan sel yang tidak terkendali akibat mutasi gen menyebabkan pembentukan hamartoma.
Gejala hamartomas
Tergantung pada lokasi hamartoma, jenisnya dibedakan, dan masing-masing memiliki struktur dan simtomatologi sendiri.
Hamartoma paru-paru
Hamartoma paru dapat terbentuk di lobus dan bagian perifer paru mana pun dan terdiri dari jaringan normal yang ada di paru-paru: jaringan adiposa, epitel, fibrosa, dan tulang rawan. Dalam 80% kasus, komponen kondroid (sel tulang rawan hialin) mendominasi dengan masuknya adiposit - sel jaringan adiposa dan sel epitel saluran napas. [ 3 ]
Nama-nama sebelumnya: hamartoma kondroid, mesenkimoma, hamartoma kondromatosa, atau hamartokondroma saat ini tidak direkomendasikan oleh WHO.
Sebaliknya, hamartoma kistik mesenkimal paru-paru kurang umum dan dikaitkan dengan sindrom Cowden pada sebagian besar pasien.
Lesi hamartomatosa pada paru mungkin tidak menunjukkan gejala, namun dapat menimbulkan gejala berupa batuk kronis (sering disertai hemoptisis), mengi saat bernafas dan kesulitan bernafas. [ 4 ]
Hamartoma pada jantung
Tumor jantung primer jinak pada orang dewasa meliputi hamartoma miosit matur, dan pada bayi dan anak-anak dengan sklerosis tuberosa, rabdomioma, yaitu hamartoma miokardium pada ventrikel atau septum interventrikular. [ 5 ]
Hamartoma kardiomiosit dewasa berkembang di dinding ventrikel (dan jarang di atrium) dan dapat muncul sebagai beberapa lesi yang merupakan massa padat yang terkait erat dengan miokardium di bawahnya. Tumor dapat menyebabkan gejala gagal jantung: nyeri dada, palpitasi dan aritmia, murmur jantung, edema, dispnea, sianosis.
Rhabdomyoma jantung, yang sebagian besar terdiagnosis dalam tahun pertama kehidupan, tersusun dari jaringan otot jantung yang dibentuk oleh mioblas embrionik dan memiliki tampilan massa fokal padat tanpa kapsul.
Biasanya, hamartoma ini muncul tanpa gejala dan akan mengalami regresi spontan sebelum usia 4 tahun.
Lesi hamartomatosa juga dianggap oleh beberapa ahli berhubungan dengan miksoma kompleks Carney pada jantung. [ 6 ]
Gamartoma pada saluran pencernaan
Hamartoma lambung adalah massa mesenkimal berupa polip hiperplastik epitel lambung, polip Peutz-Jeghers, dan hamartoma mioepitel langka - dengan berkas otot polos yang mengalami hipertrofi. Nama lain untuk hamartoma ini meliputi hamartoma mioglandular, hamartoma adenomiomatosa, dan adenomioma lambung. Manifestasi klinis yang umum meliputi dispepsia, nyeri epigastrik, dan perdarahan GI bagian atas. [ 7 ], [ 8 ]
Informasi lebih lanjut dalam materi - Poliposis Lambung
Hamartoma intestinal adalah polip hamartomatosa atau hiperplastik pada usus besar, yang didiagnosis sebagai adenoma adenomatosa atau tubular. Bila hamartoma terlokalisasi di kelenjar Brunner duodenum, gejalanya berupa nyeri di daerah epigastrik; mual, muntah, dan perut kembung (menunjukkan obstruksi usus); dan, bila ukurannya cukup besar, perdarahan gastrointestinal. Pada kasus hamartoma mioepitelial ileum, pasien mengeluhkan nyeri perut, berat badan menurun, dan mengalami anemia kronis. [ 9 ], [ 10 ]
Baca juga - Polip Rektal
Hamartoma retrorektal adalah hamartoma kistik atau kista multibilik pada ruang retrorektal (jaringan ikat longgar antara rektum dan fasianya sendiri) yang paling sering terjadi pada wanita paruh baya. Bentuknya seperti kista yang menonjol keluar dari dinding posterior rektum, yang dilapisi epitel dan mengandung serat otot polos yang tersusun tidak beraturan. Hamartoma ini disertai nyeri perut bagian bawah dan konstipasi berulang. [ 11 ], [ 12 ]
Hamartoma pada hati dan limpa
Hamartoma bilier multipel pada hati adalah hamartoma pada duktus biliaris intrahepatik interdigitalis yang terkait dengan malformasi perkembangannya selama periode embrionik. Hamartoma ini (tunggal atau multipel) terdiri dari kelompok duktus biliaris yang melebar secara acak dan stroma fibrokolagen. [ 13 ]
Hamartoma bilier tidak bergejala dan biasanya ditemukan secara tidak sengaja (selama pemeriksaan radiologi atau laparotomi). [ 14 ]
Neoplasma primer jinak yang jarang dan sering terdeteksi secara tidak sengaja adalah hamartoma limpa, yang terdiri dari unsur-unsur pulpa merah limpa - dalam bentuk massa homogen yang jelas dengan konsistensi yang kuat. Malformasi ini bisa tunggal atau ganda; saat meremas parenkim limpa, mungkin ada perasaan tidak nyaman dan nyeri di daerah subkostal kiri. [ 15 ], [ 16 ]
Hamartoma ginjal
Hamartoma ginjal yang paling umum didiagnosis sebagai angiomyolipoma ginjal, karena tumor jinak ini terdiri dari jaringan adiposa matang dengan serat otot polos dan pembuluh darah yang tertanam. Tumor ini terbentuk pada sklerosis tuberosa dalam 40-80% kasus. Peningkatan ukuran hamartoma (lebih dari 4-5 cm) menyebabkan rasa sakit dan munculnya darah dalam urin. [ 17 ], [ 18 ]
Hamartoma payudara
Definisi diagnostik hamartoma payudara yang diterima WHO adalah istilah-istilah seperti adenolipoma, kondrolipoma, dan hamartoma mioid. Meskipun sering disebut fibroadenolipoma oleh ahli mammologi, karena pembentukan tumor mengandung sel-sel jaringan fibrosa, kelenjar, dan adiposa yang tertutup dalam kapsul jaringan ikat tipis dengan garis luar yang jelas. Kalsifikasi fokal dapat diamati saat visualisasi. Dalam kasus ini, manifestasi klinis tidak ada. [ 19 ], [ 20 ]
Baca juga - Tumor Payudara
Hamartoma otak
Sepertiga pasien dengan sklerosis tuberosa memiliki hamartoma otak dalam bentuk pertumbuhan kortikal intrakranial atau tuberkel di berbagai lobus - di perbatasan materi abu-abu dan putih - atau nodul subependimal di sepanjang dinding ventrikel otak. Hamartoma astrositik, astrositoma sel raksasa subependimal dengan gangguan kortikal, neuron dismorfik, dan sel glia besar parenkim otak (astrosit), juga dapat terbentuk. Gejala hamartoma serebral meliputi serangan kejang dan keterbelakangan mental pada anak-anak. [ 21 ], [ 22 ]
Malformasi langka yang terjadi selama embriogenesis dan sudah ada sejak lahir adalah hamartoma hipotalamus, yang merupakan massa neuron heterotopik dan sel glia. Seiring pertumbuhan otak anak, tumor membesar tetapi tidak menyebar ke daerah otak lainnya. [ 23 ], [ 24 ]
Bila jaringan hipertrofi terbentuk di bagian anterior hipotalamus (tuber cinereum), tempat kelenjar pituitari menempel padanya, malformasi tersebut memperlihatkan gejala perkembangan seksual prematur sentral (sebelum usia 8-9 tahun): munculnya ruam jerawat, perkembangan dini kelenjar susu, dan menarche dini pada anak perempuan; rambut kemaluan dan mutasi suara dini pada anak laki-laki.
Bila hamartoma terbentuk di bagian posterior hipotalamus, mungkin ada kelainan pada aktivitas listrik otak, yang pada awal masa bayi dimanifestasikan oleh kejang, dan pada tahap selanjutnya (usia 4 sampai 7 tahun) oleh epilepsi dengan kejang epilepsi fokal disertai tawa tiba-tiba atau tangisan tak terkendali, kejang atonik dan tonik-klonik, serta kejang agresi, memori, dan masalah kognitif.
Hamartoma pituitari merupakan adenoma pituitari jinak yang terjadi secara sporadis.
Orang dewasa setengah baya dengan sindrom Cowden mungkin memiliki massa mirip tumor langka, hamartoma serebelum, yang didiagnosis sebagai gangliocytoma serebelum displastik atau penyakit Lhermitte-Duclos. Gejala mungkin tidak ada atau bermanifestasi sebagai sakit kepala, pusing, gangguan koordinasi gerakan, dan kelumpuhan saraf kranial individu.
Hamartoma kelenjar getah bening
Bila sel otot polos dan jaringan adiposa, serta pembuluh darah dan stroma kolagen pada kelenjar getah bening inguinal, retroperitoneal, submandibular dan serviks tumbuh berlebihan, maka akan terbentuk hamartoma angiomioma pada kelenjar getah bening atau hamartoma angiomioma nodular - dengan penggantian parenkimnya sebagian atau seluruhnya. [ 25 ], [ 26 ]
Hamartoma pada kulit
Bila terjadi sklerosis tuberosa atau neurofibromatosis, berbagai hamartoma kulit teramati, paling sering berupa bercak hipopigmentasi; bercak kopi dan susu; angiofibroma (pada pipi, dagu, lipatan nasolabial); bercak shagreen pada berbagai lokasi (yang merupakan nevi jaringan ikat); plak fibrosa pada dahi, kulit kepala, atau leher.
Manifestasi dermatologis langka dari tuberous sclerosis (terutama pada pria) adalah hamartoma folikulosis dan kolagen, ditandai dengan penumpukan kolagen yang melimpah di dermis, fibrosis perifolikular konsentris, dan kista subkutan berbentuk corong berisi keratin yang terlihat pada pemeriksaan histopatologi. [ 27 ]
Untuk hamartoma yang terdiri dari melanosit (sel yang menghasilkan pigmen melanin), sebagian besar ahli juga merujuk pada berbagai neoplasma melanositik, khususnya nevi melanositik kongenital, yang merupakan kelainan embriogenesis.
Dari segi etiologi, hamartoma yang terdiri dari jaringan vaskular juga merupakan hemangioma kulit.
Penderita sindrom Peutz-Jeghers-Thuren memiliki hamartoma berupa bercak-bercak pigmentasi pada kulit dan selaput lendir - lentiginosis periorificialis
Kasus hamartoma ektodermal-mesodermal papular linear (Hamartoma moniliformis) menunjukkan ruam papular berwarna daging linear pada kepala, leher, dan dada bagian atas.
Dan hamartoma sebositik adalah hamartoma pada kelenjar sebasea, baca selengkapnya dalam publikasi - nevus sebasea.
Hamartoma pada mata
Lesi hamartomatosa berpigmen pada iris pada neurofibromatosis tipe 1 dan sindrom Watson - dalam bentuk gugus nodular melanosit dendritik - didefinisikan sebagai hamartoma iris atau nodul Lisch. Lesi ini berupa papula kuning-coklat berbentuk kubah yang transparan (biasanya tidak mengganggu penglihatan) yang menonjol di atas permukaan iris.
Dan pasien dengan angiofibroma juvenil nasofaring dan poliposis adenomatosa familial sering mengembangkan hamartoma gabungan retina dan epitel pigmen retina - dalam bentuk bintik hitam di bagian tengah (makula) retina. [ 28 ]
Hamartoma hidung
Hamartoma nasal didefinisikan oleh para spesialis sebagai hamartoma kondromesenkim nasal atau kondroma nasal, yang disebabkan oleh proliferasi jinak epitel pernapasan, kelenjar submukosa, dan mesenkim tulang kondro. Manifestasi klinisnya bergantung pada ukuran dan lokasi lesi dan meliputi: hidung tersumbat, kesulitan bernapas melalui hidung dan menyusui pada bayi, keluarnya cairan bening encer dari hidung, dan mimisan. Hamartoma dapat tumbuh bersama anak dan menyebar ke dalam rongga mata, yang mengakibatkan perpindahan bola mata ke depan atau ke belakang, strabismus, atau gangguan okulomotor. [ 29 ]
Hamartoma pada anak
Semua lesi hamartomatosa berbagai organ dan struktur anatomi yang disebutkan di atas terdapat pada anak-anak dengan sindrom yang sesuai.
Bayi baru lahir memiliki hamartoma mesenkimal pada dinding dada atau hamartoma tulang rawan pada tulang rusuk, yang merupakan massa padat tak bergerak yang dihasilkan dari pertumbuhan berlebih fokal elemen rangka normal dengan elemen tulang rawan, pembuluh darah, dan mesenkimal. Hamartoma ini dapat menyebabkan kegagalan pernapasan dan perkembangan sindrom gangguan pernapasan. Hamartoma mesenkimal hati adalah tumor hati jinak kedua yang paling sering terjadi pada anak-anak. Pembentukan seperti tumor ini (lebih sering terlokalisasi di lobus kanan organ) terdiri dari sel-sel stroma mesenkimal, hepatosit, dan sel epitel lapisan saluran empedu. Gambaran klinisnya meliputi massa yang teraba di rongga perut, anoreksia, dan penurunan berat badan, dan jika ukurannya signifikan (hingga 10 cm dan lebih), tumor menutupi saluran empedu ekstrahepatik dan vena cava inferior, yang menyebabkan penyakit kuning dan edema pada ekstremitas bawah.
Hamartoma adalah nefroma mesoblastik kongenital (terjadi pada 1 dari 200.000 bayi) yang dapat menyebabkan perut kembung pada bayi baru lahir dengan massa padat yang teraba di kuadran kanan atas perut. Bayi juga dapat mengalami pernapasan cepat dan dangkal.
Kelainan kongenital yang langka meliputi hamartoma fibrosa pada bayi, yang terjadi pada anak-anak dalam dua tahun pertama kehidupan dan muncul sebagai massa nodular yang tidak nyeri pada jaringan subkutan aksila, leher, bahu dan lengan bawah, punggung dan dada, paha, kaki, dan genitalia luar.
Hamartoma angiomatosa ekrin pada anak dapat muncul sejak lahir atau muncul pada awal masa kanak-kanak. Tumor jinak yang bersifat hamartomatosa ini biasanya tampak seperti nodul dan/atau plak kebiruan atau kecokelatan yang disebabkan oleh proliferasi jaringan kelenjar keringat ekrin dan kapiler di lapisan tengah dan dalam dermis. Hamartoma ini dapat menyebabkan hiperhidrosis lokal dan peningkatan pertumbuhan rambut.
Komplikasi dan konsekuensinya
Secara umum disepakati bahwa hamartoma jarang kambuh atau berubah menjadi tumor ganas. Hamartoma sering kali menunjukkan sedikit atau tidak ada gejala dan terkadang bahkan menghilang seiring berjalannya waktu. Namun, pada kasus yang lebih parah dan tergantung pada lokasi pembentukannya, malformasi ini dapat menimbulkan komplikasi dan konsekuensi serius.
Pertama-tama, hamartoma dapat tumbuh sedemikian besarnya sehingga mulai menekan jaringan dan organ di sekitarnya, sehingga mengganggu fungsinya.
Hamartoma jantung pada anak-anak dapat menyebabkan kelainan irama jantung persisten, cacat katup, dan gangguan aliran darah intrakardiak dengan gagal jantung kongestif berikutnya.
Komplikasi polip hamartomatosa pada saluran cerna adalah pendarahan gastrointestinal, obstruksi, dan intususepsi usus (dengan kemungkinan kematian). Dan hamartoma ginjal yang besar dapat memicu ruptur ginjal.
Hamartoma di otak dapat menyebabkan sindrom hidrosefalus obstruktif.
Pada hamartoma hipotalamus dan pituitari, produksi hormon somatotropik (hormon pertumbuhan) dapat terganggu, yang menyebabkan perkembangan nanisme hipofisis (hipopituitarisme) pada anak-anak. Hamartoma hipotalamus pada anak-anak juga dapat menyebabkan epilepsi yang resistan terhadap obat.
Komplikasi hamartoma epitel pigmen retina penuh dengan disfungsi retina dan/atau saraf optik, edema makula, neovaskularisasi koroid, dan ablasi retina.
Diagnostik hamartomas
Bagian penting dari diagnosis hamartoma dan sindrom terkait adalah pengumpulan anamnesis, termasuk riwayat keluarga.
Pemeriksaan laboratorium meliputi pemeriksaan darah: klinis umum; elektrolit serum; profil limfosit; kadar kalsium, kalium, fosfat, dan urea; serta pemeriksaan fungsi hati. Jika memungkinkan, biopsi aspirasi jarum halus pada massa dilakukan, karena pemeriksaan histologis sangat penting dalam diagnosis dan pemilihan taktik pengobatan.
Diagnostik instrumental memberikan visualisasi pembentukan mirip tumor hamartomatosa dan identifikasi lokalisasi pastinya, yang mana digunakan sinar X, angiografi, elektroensefalografi (EEG), ultrasonografi (sonografi), CT (computed tomography), PET (positron emission tomography), MRI (magnetic resonance imaging).
Perbedaan diagnosa
Pada setiap massa abnormal, diagnosis banding sangatlah penting. Dengan demikian, tuberkuloma dan hamartoma dibedakan; hamartoma paru dan kanker paru primer, karsinoid bronkogenik, penyakit metastasis. Hamartoma otak harus dibedakan dari kraniofaringioma dan glioma hipotalamus-kiasmatik. Dan diagnosis banding hamartoma sebagai nefroma mesoblastik kongenital meliputi tumor Wilms (nefroblastoma ganas), sarkoma sel jernih pada ginjal, dan tumor ginjal yang mengeras pada bayi.
Siapa yang harus dihubungi?
Pengobatan hamartomas
Jika hamartoma tidak bergejala dan ditemukan secara tidak sengaja, tidak diperlukan pengobatan, tetapi perlu untuk memantau "perilakunya" dan kondisi pasien. Dalam kasus lain, terapi ditujukan untuk mengurangi intensitas gejala dan mencegah komplikasi. Misalnya, pada hamartoma hipotalamus dengan gejala pubertas dini, obat-obatan tertentu yang menghambat pelepasan hormon tertentu diresepkan. Obat-obatan jantung digunakan untuk mengobati gejala gagal jantung pada pasien dengan hamartoma jantung.
Operasi pengangkatan hamartoma diindikasikan untuk memastikan diagnosis dan dalam kasus gejala parah yang tidak dapat diperbaiki secara medis.
Misalnya, hamartoma paru-paru dapat diangkat dengan reseksi baji dan, pada kasus yang parah, dengan pengangkatan lobus paru-paru (lobektomi). Hamartoma payudara juga dapat diangkat, dan jika ukurannya besar, mastektomi parsial atau komplet mungkin diperlukan.
Termoablasi frekuensi radio stereotaktik atau ablasi laser dapat digunakan untuk mengangkat polip hamartomatosa. Bedah radio dengan sinar gamma yang sangat terfokus - pisau gamma untuk hamartoma hipotalamus atau hamartoma astrositik - juga digunakan.
Pencegahan
Satu-satunya metode pencegahan perkembangan hamartoma dapat dipertimbangkan melalui pemeriksaan genetik calon orang tua anak tersebut.
Ramalan cuaca
Prognosis keseluruhan dari anomali kongenital ini bergantung pada lokasi dan ukuran neoplasma, serta pada penyakit penyerta dan kesehatan umum pasien.